Common diseases of adult life such as diabetes mellitus, hypertension, schizophrenia, alcoholism
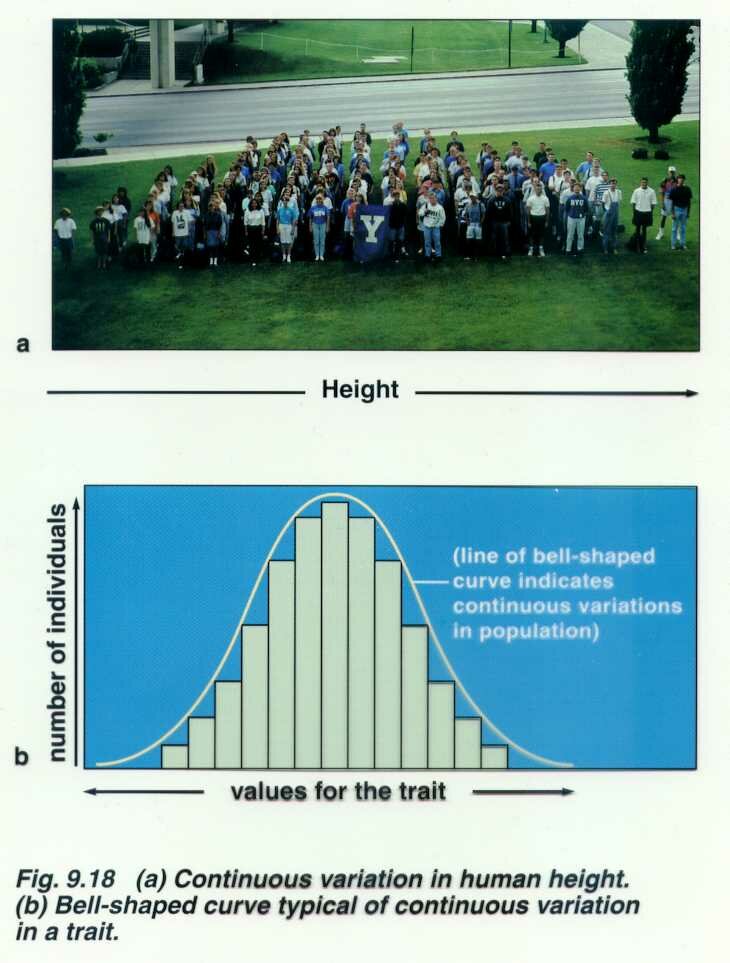
Normal variation in height, skin color, IQ
MULTIFACTORIAL AND COMPLEX INHERITANCE
Although many human disorders are inherited as single gene defects or as chromosome abnormalities, many common diseases and common congenital malformations are not. It is thought that for many disorders that several pairs of additive genes are involved. These traits are referred to as polygenic (several pairs of genes at different loci with additive effects) or multifactorial which means they are polygenic but also influenced by other genes and the pre and post natal environment. Fingerprint ridge count appears to be purely polygenic, height and skin color are multifactorial. Height is influenced by the sex of the individual and by nutrition.
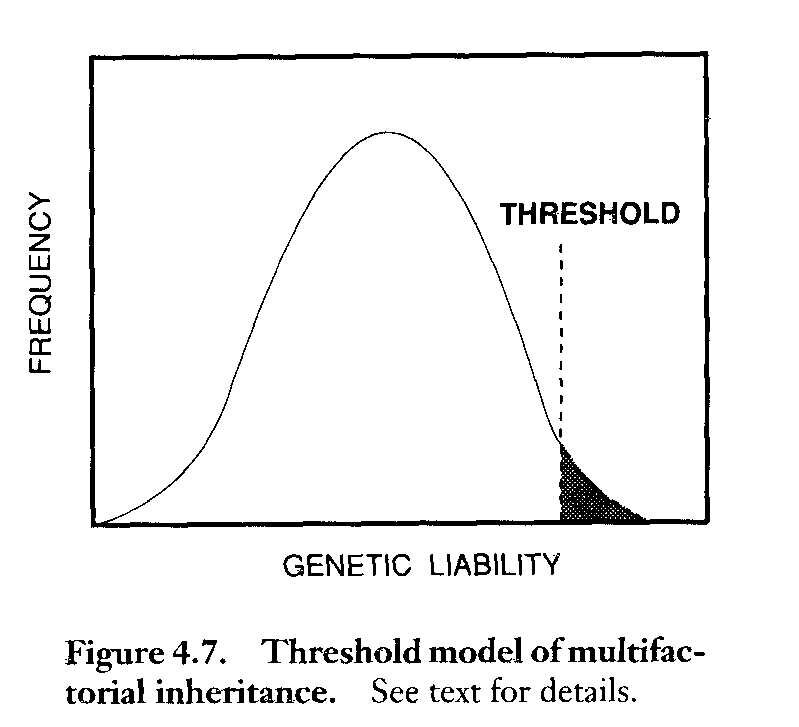
Some multifactorial traits are often expressed more frequently in one or the other sex. For example, cleft lip/cleft palate is more common in males. One subgroup of multifactorial traits, such as CL/CP, exhibit a threshold effect. This refers to the hypothesis that one must have a certain number of genes for the trait to express. In the case of CL/CP female have a higher threshold and must have more genes to express the trait. Therefore, an affected female has a higher risk than an affected male of having an affected male child. All people are assumed to carry some of these genes so both parents contribute genes. The following page is a list of several traits considered at this time to be multifactorial. Neural tube defects (NTDs) are in this category. NTDs include anencephaly (lethal) and spina bifida (causes paralysis). NTDs are more frequent in Hispanics. It has been found that high levels of folate (folic acid) can reduce the risk of having a child with an NTD. It also reduces the risk of other birth defects and protects adults from heart disease.
More recently the term, oligogenic, has been used for traits with a major susceptibility locus but whose expression is also influenced by a few other genes as well as the environment. Diabetes, hypertension, cancer, schizophrenia are examples of oligogenic traits. Often the use of the terms multifactorial and oligogenic are more a reflection of our current lack of knowledge about the real genetic basis of the disorder than an accurate description of an inheritance pattern. The genetics of common disease is only recently being explored. In many of these disorders, there are major susceptibility loci but they differ in different families, Alzheimer, asthma, schizophrenia are examples.
SOME EMPIRIC RISKS FOR COMMON (MULTIFACTORIAL) CONGENITAL ANOMALIES
In general, if 2 unaffected parents have 2 affected children, the risk doubles, or it the affected parent has already had 1 child, the risk also doubles
| Normal parents 1 affected child subsequent children |
1 affected parent risk for first child |
Identical Twin |
Male/Female Ratio |
Population Incidence |
|
| Cleft lip and palate |
4% 2.5% unilateral lip 5.6% bilateral lip/palate |
3.2% | 31% | 2:1 | 1/1000 |
| whites | 1/750 | ||||
| blacks | 1/2500 | ||||
| Navajos | 1/500 | ||||
| Japanese | 1/600 | ||||
| B. C. Indians | 1/380 | ||||
| Cleft palate | 2% | 6% | 40% | 3:2 | 0.4/1000 |
| whites | 1/2000 | ||||
| blacks | 1/2500 | ||||
| Navajos | 1/2800 | ||||
| Clubfoot | 3% | 3% | 33% | 2:1 | 1.2/1000 |
| Congenital heart disease | |||||
| VSD | 4 - 5% | 3 - 4% | 1.3:1 | 5/1000 | |
| PDA | 1 - 4% | 2.8% | 1/2000 | ||
| Tetralogy of Fallot | 2 - 3% | 1.6% | |||
| ASD | 3% | 3.5% | 1/1000 | ||
| Pulmonary stenosis | 3% | 3% | |||
| Aortic stenosis | 3% | ||||
| Coarctation | 2% | ||||
| Transposition | 2% | ||||
| AV Canal | 2 - 3% | ||||
| More complex anomalies |
1 - 2% | ||||
| NTD spina bifida anencephaly |
2% B.C. 3% U.S.A. 5% G.B. |
2%
3% |
21% |
1/700
1/330 London |
|
| whites Jews blacks Puerto Ricans |
1/700 Boston 1/1200 1/1500 1/500 |
||||
| Congenital hip dislocation | 3.5% | 3 - 5% | 35% | 1:7 | 2/1000 |
| Pyloric stenosis | 3.2% if brother affected 6.5% if sister affected |
25.4% if mother affected 4.2% if father affected |
> males affected |
(The following was taken from a Power Point presentation on Multifactorial Inheritance but does not include some of the figures and tables.)
Multifactorial Inheritance and Complex Traits
Almost all disorders in (hu)man(s) are familial in that they are more likely to afflict someone with an affected relative than someone with an equivalent set of unaffected relatives. J H Edwards in BR Med Bull 1969;25:58-64
Multifactorial and complex traits include:
Common diseases of adult life such as diabetes mellitus, hypertension, schizophrenia, alcoholism
Normal variation in height, skin color, IQ
Common congenital malformations such as cleft lip, cleft palate, neural tube defects
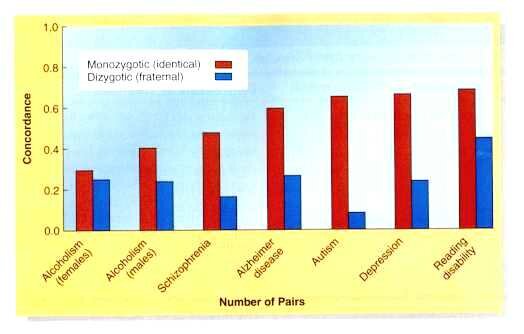
To tease apart genetic and environmental factors of complex traits, geneticists turn to DZ and MZ twins
For DZ and MZ twins: a trait with a greater concordance between MZ twin pairs than DZ twin pairs is at least partially controlled by heredity
They also study MZ twins separated at birth since many of their traits held in common can be attributed to genetics, especially if their environments have been very different.
Their differences reflect the effect of environment.

Comparison of adoptees with their biological and adoptive parents provide information
Similarities between adopted people and adoptive parents reflect environmental influences.
Similarities between adoptees and their biological parents mostly reflect genetic influences.
Examples include alcoholism studies
Alcoholism among male adoptees (table in presentation)
| Biological Parent | # in sample | % of adopted sons who were alcoholic | averages of both parents |
| Alcoholic father | 89 | 39.4 | 34% |
| Alcoholic mother | 42 | 28.6 | |
| Non alcoholic father | 723 | 13.6 | 14.6% |
| Non alcoholic mother | 1029 | 15.5 |
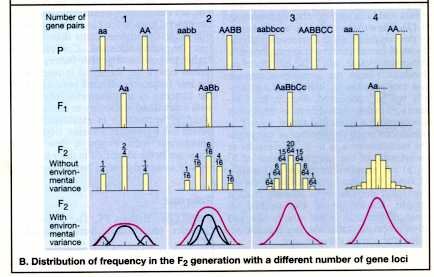
Polygenic Traits
Are quantitative rather than qualitative in nature. They are frequently distributed continuously in the population, often in a more or less bell shaped curve or they may fit the threshold model
Polygenic traits include height, blood pressure, cleft lip ± cleft palate, NTDs
A frequency distribution of systolic blood pressure determined by a two-locus two allele model was presented.
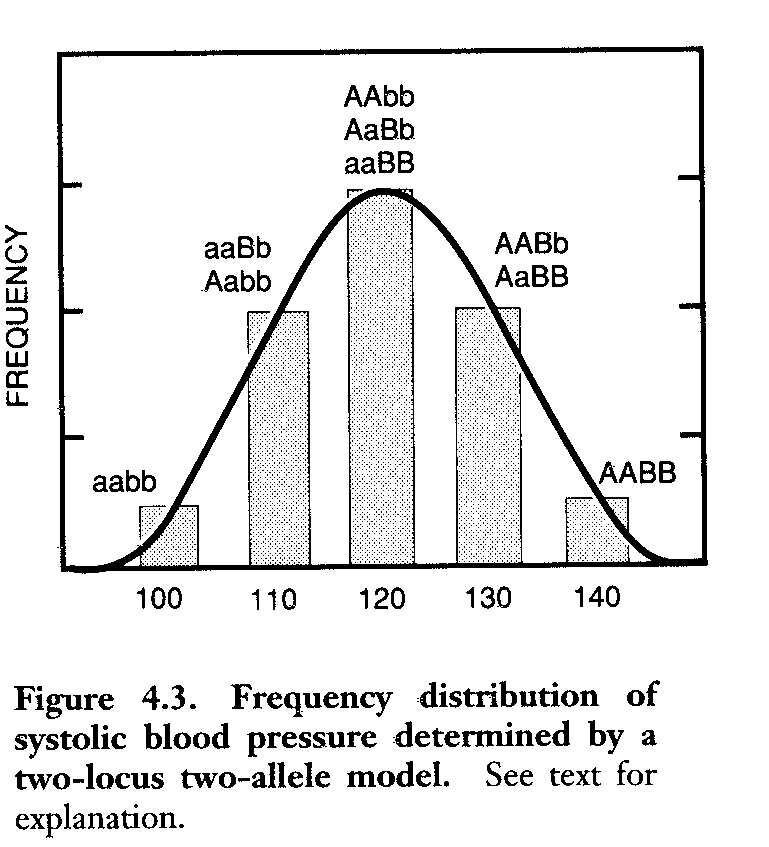
Also a model showing the effect of different numbers of gene loci on the phenotypic frequency distribution
The Threshold Model:
Discontinuous traits from continuously distributed liability
Recurrence Risks for Polygenic Disease or Malformation
Risks are different from Mendelian inheritance risks
Risks represent average risks and will vary among different families
The risk increases with the number of affected relatives
The risk increases with the severity of the malformation or disease
The differential risk to relatives of an affected proband increases as the frequency of the disease or malformation in the general population decreases.
When the sex ratio of affected probands deviates significantly from unity, offspring of affected probands of the less frequently affected sex are at higher relative risk.
Terminology used for relatives of the proband depends on the number of shared genes
First-degree: parents, sibs and offspring of the proband share 50% (1/2) of their genes
Second-degree: grandparents and grandchildren, uncles and aunts, nephews and nieces, half-sibs share 25% (1/4) of their genes
Third-degree: first cousins, great-grandparents, great grandchildren share 12.5% (1/8) of their genes
Proportion of Children Affected with Pyloric Stenosis
n Proband Children
Sons Daughters
Father 5.5 % 2.4%
Mother 19.4 % 7.3%
Population 0.5% 0.1%
Incidence
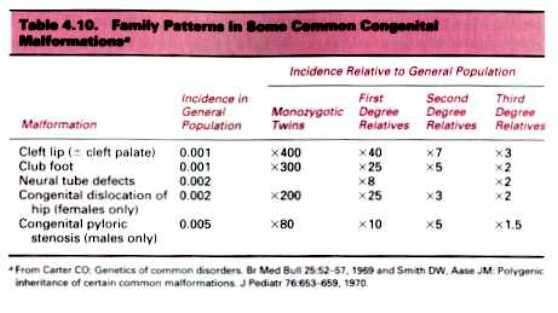
Family patterns in some common congenital malformations (given in presentation)
Heritability = G = G
G + B + E (total phenotypic variance) V
= variance in DZ pairs-variance in MZ pairs
variance in DZ pairs
B (family environment) E (random environmental factors)
Many multifactorial traits are influenced both by genes and environment
The concept of heritability tries to separate their relative roles.
By definition, heritability is the proportion of the total phenotypic variance (V) of a trait that is caused by additive genetic variance.
Summary of the characteristics of multifactorial inheritance
1. Although the disorder is obviously familial, there is no distinctive pattern of inheritance within a single family
2. The risk to first-degree relatives, determined from family studies, is approximately the square root of the population risk
3. The risk is sharply lower for second-degree than for first-degree relatives, but it declines less rapidly for more remote relatives.
4. The recurrence risk is higher when more than one family member is affected.
5. The more severe the malformation, the greater the recurrence risk.
6. If a multifactorial trait is more frequent in one sex than in the other, the risk is higher for relatives of patients of the less susceptible sex.
7. If the concordance rate in DZ twins is less than half the rate in MZ twins, the trait cannot be autosomal dominant, and if it is less than a quarter of the MZ rate, it cannot be autosomal recessive.
8. An increased recurrence risk when the parents are consanguineous suggests that multiple factors with additive effects may be involved.
HUMAN DISEASE CATEGORIES. While these categories served practical purposes such as recurrence risk assessment, there are many overlaps among them.
Here is another group of definitions from Sheffield, V. et al. (1998) Trends Genet.14, 391-396, with some modifications by CDG.
Chromosomal disorders Disorders that result from an abnormality in the number of chromosomes, or that result from the rearrangement, duplication or deletion of large chromosomal regions.
Monogenic disorders Diseases that result from mutation in a single gene. Complexity with this group arises from the fact that mutations in different individual genes often result in clinically indistinguishable phenotypes (locus heterogeneity). Another complexity is that of variable expressivity and incomplete penetrance. Even purely monogenic disorders can be modified by other genes and/or environmental factors. An example is PKU control by lowering phenylalanine in the diet. Mitochondrial disorders could be placed in this category.
Polygenic disorders Diseases that arise when mutations in more than one gene contribute to the disease. The simplest are those that have a single major genetic component modified by one or a few modifier genes. An example is a susceptibility locus for congenital heart disease that was mapped to chromosome 1. The locus shows incomplete penetrance and variable expressivity indicating that other genes and/or environmental factors influence the phenotype. There can also be interaction of multiple genetic mutation, each of which contribute to the phenotype. Each mutant gene could contribute to the disorder in an additive fashion, leading to a quantitative phenotype or contribute to reaching a disease threshold.
Multifactorial disorders Phenotypes that result from the interaction of genetic and environmental components.
Environmental disorders Diseases that result principally from environmental influences. Even disorders of this type can have genetic susceptibility components. An example is resistance to HIV-1 infections in individuals with variant alleles of the CCR5 and CCR2 chemokine receptor genes.
TWENTY-FIVE MOST COMMON MULTIPLE CONGENITAL ANOMALY SYNDROMES
767 Cases seen at UCSF Genetic Service 1970 -1980
| MALFORMATION SYNDROME | # OF CASES | ETIOLOGY |
| Trisomy 21 (Downs Syndrome) | 328 | Chromosomal |
| Potter Oligohydramnios Sequence | 38 | Unknown; Sporadic; recurrence risk for sibs 1-3% |
| Amniotic Band Syndrome | 34 | Unknown; Sporadic; low recurrence risk (AD/AR?) |
| Osteogenesis Imperfecta | 30 | Single Gene (AD) |
| Trisomy 18 Edward Syndrome | 30 | Chromosomal |
| VACTERL Association | 26 | Unknown; Sporadic; RR 1%; more frequent in diabetic mothers (AD/mt?) |
| Marfan Syndrome | 23 | Single Gene (AD) |
| Prader-Willi | 22 | Microdeletion; UPD; Imprinting; 1.6 % RR |
| Noonan Syndrome | 21 | Single Gene (AD) (some families); Unknown/Sporadic in others |
| Williams Syndrome | 18 | Microdeletion, Sporadic; Low RR |
| Achondroplasia | 17 | Single Gene (AD) |
| Trisomy 13 Patau Syndrome | 16 | Chromosomal |
| Turners Syndrome | 16 | Chromosomal |
| Ehlers-Danlos Syndrome | 16 | Single Gene (AD) |
| Rubinstein-Taybi Syndrome | 16 | Deletion; Sporadic; Low RR (AD?) |
| Klippel-Trenaunay-Weber Syndrome | 15 | Unknown; Sporadic; Low RR (AD?) |
| Fetal Alcohol Syndrome | 14 | Teratogen |
| de Lange Syndrome | 14 | Unknown; Sporadic; Recurrence Risk for sibs 2% (AD) |
| Moebius Syndrome | 14 | Unknown; sporadic; RR 1-2% |
| Hemifacial Microsomia (Goldenhar Syndrome) | 13 | Unknown; Sporadic; Low RR (AD?) |
| Pierre Robin Sequence | 10 | Unknown; Sporadic; Low RR (AD?) |
| CHARGE Association | 10 | Unknown; Sporadic; Low RR (AR/XL/multifactorial?) |
| Laurence-Moon-(Biedl-Bardet) Syndrome | 9 | Single Gene (AR) |
| Russel-Silver Syndrome | 9 | Unknown; Sporadic; Low RR (AD/XL) |
| Fetal Dilantin Syndrome | 9 | Teratogen |
PERCENTAGES OF CASES IN MAJOR MULTIPLE CONGENITAL ANOMALIES ETIOLOGIC CATEGORIES
| ETIOLOGIC CATEGORY | NUMBER OF CASES |
PERCENTAGE %
|
| Chromosomal |
428
|
66.56%
|
| Single Gene |
9
|
1.40%
|
| Teratogen |
23
|
3.58%
|
| Unknown; Sporadic (Mixture) |
183
|
28.46%
|
| Total |
767
|
100.00%
|
SLIDE SHOW OF AD, AR, XR AND MULTIFACTORIAL TRAITS.
We have covered some of these previously and will cover others in future lectures. However, this is your cue to look them up in your textbooks and learn about their etiologies. I will show these in class or you can check them out and look at them.
The first slide is of Mendel who is considered the father of genetics who became an Abbey of his monastery for reasons we can only guess. He did his work around 1865 and was rediscovered by three independent researchers around the turn of the century. Mendel did not coin the word "genes", Bateson did, he used the term "factors." He also did not know about chromosomes nor did he know about linkage. All of the traits he studied were either on different chromosomes or so far apart on the same chromosome that they assorted independently. He may have found his law of independent assortment did not work for all two factor crosses (if they were linked) and gave up and became an administrator. Bateson was the first one to describe linkage. In human genetics we rarely work with more than one pair of genes at a time so we, like Mendel, need not concern ourselves with linkage very often.
Achondroplasia is an AD trait that is fully penetrant. It is the most frequent form of short-limb dwarfism. About 7/8 of cases are the result of new mutation, there being a considerable reduction of effective reproductive fitness. There is a paternal age effect. Gonadal mosaicism (or spermatogonial mutation) is a possible explanation for the occasional report of affected sibs from normal parents. Affected individuals have rhizomelic shortening of the limbs, a characteristic facies with frontal bossing and mid-face hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum (bow legs), and trident hand. The mutation is in a gene coding for a fibroblast growth factor receptor (FGFR-3) located on chromosome 4 at p16.3. People with AD traits if they are rare are heterozygous and rarely does one get to see the homozygous phenotype. However, since individuals with achondroplasia meet in organizations such as Little People of America, there have been mating. When this occurs about 25% (overall) of the offspring are severely affected and often die in utero. Therefore, the trait is not a true AD since two doses of the mutant gene is obviously not equal in severity to one. 25% of the offspring of a mating between heterozygotes will be of normal stature and 50% will have achondroplasia like their parents. Usually the matings are between heterozygotes and homozygous normal individuals, in which case the probability of having a child with normal stature or with achondroplasia is equal (50%).
Crouzon syndrome is an AD craniofacial dysostosis also due to a mutation in a fibroblast growth factor receptor (FGFR-2) on chromosome 10. There is craniosynostosis, the eyes have shallow orbits, there is proptosis, hypertelorism, parrot-beaked nose, short upper lip, hypoplastic maxilla and mandibular prognathism.
Abraham Lincoln is believed to have had the AD Marfan syndrome but some question this. However, Paganini, the great violinist is believed by many to have had it. And, of course, the great athlete, Flo Hyman, died of the consequences of Marfan. This disorder is caused by a defect in fibrillin a connective tissue component. It is characterized by above average and disproportionate height, arachnodactyly, dislocation of the lens, joint laxity, striae, and scoliosis or kyphosis. It is sometimes treated with propanolol in an attempt to prevent aneurysms. Some basketball players with Marfan syndrome have died from it. Pregnant women with the syndrome need special care due to the stress put on the body.
Treacher Collins is an AD with variable expressivity. You saw a picture of a father and son. The son was much more severely affected and the father who only showed mild expression of the disorder was only diagnosed after the birth of his son. The phenotype can include a reduced mandible, malformed ears, deafness, cleft palate and mental handicap. The gene (TCOF1) is at 5q32-q33.1 and codes for a protein of unknown function called TREACLE.
Neurofibromatosis (NF1) is caused by the inheritance of a mutant tumor suppressor gene and the subsequent loss of heterozygosity for the gene in the affected tissues. The gene product is called neurofibromin. NF1 shows extreme variation in expression since the neurofibromas show up in a wide variety of locations in the body. If they are present in the brain they can cause mild to severe mental retardation. One manifestation is the presence of multiple café au lait spots on the body especially in the axillary region. The gene is extremely large and many cases are due to new mutations. Since mutations are known to occur at many sites within the gene, DNA analysis is difficult and instead a test is done to look for the truncated protein which is produced by the mutant gene (see handout).
Huntington disease is a true AD. It is caused by a TNR in the huntingtin gene. The function of the protein is unknown but it is widely expressed cytoplasmically. The mutant protein has a long stretch of glutamine residues which causes the protein to aggregate with other proteins. The folksinger, Woody Gurthrie, had the disorder and his son, Arlo, has a 50% risk of having inherited the mutation. Last seen, he was okay at about 50 years of age. The gene is found in a relatively high frequency with a small community in Venezuela. This is an example of a Founder effect where the gene was brought into the community by a European sailor who had many progeny. Since the community is small, there is a higher level of inbreeding and thus there are homozygotes known for the disorder but they do not manifest the disorder any differently than the heterozygotes. Thus the disorder is a "true" dominant. DNA testing for the trinucleotide expansion is available. People can now know ahead of time if they carry the disorder. This has lead to interesting ethical dilemmas when one member of a family wants to know their status and the parent (or child) does not. Also, there is some question as to the age at which a person can make an informed choice to know. Anyway, testing can be a problem in these families.
(Cornelia) de Lange, thanatophoric dwarfism and progeria are examples of new sporadic AD mutations which are also genetic lethals. TD is a perinatal lethal. Cornelia de Lange syndrome includes severe mental retardation, synophrys, hirsutism and limb deformaties.
Progeria results in premature aging with most affected persons dying in their teens. It does not affect mental abilities.
Freckles show an autosomal dominant pattern of inheritance but there is evidence of gene dosage effect as for achondroplasia and familial hypercholesterolemia. In other words, the homozygote may have more freckles than a heterozygote. By the way, freckles are found in all ethnic groups.
Retinoblastoma, like NF1, is caused by the inheritance of a mutation in a tumor suppressor gene. The cancer is caused by a loss of heterozygosity and occurs in early childhood. The retinoblastoma is usually bilateral and of early onset if inherited.
Epidermolysis bullosa results in blisters following minor trauma to the skin. EB is a group of hereditary diseases of the skin and both AD and AR patterns have been proposed. In the simplex (heals without scarring) type, epidermal basal cells are fragile and mutations of genes encoding keratin intermediate filament proteins underlie that fragility. In the dystrophic types, the causative mutations (both AD and AR) appear to be in the gene encoding type VII collagen, which is the major component of anchoring fibrils. These findings provide evidence that the cytoskeletal intermediate filament network is necessary for mechanical stability and that anchoring fibrils anchor the epidermis to the underlying dermis.
Postaxial polydactyly type A1 (PAP-A) is an AD trait with wide variation in how many hands and feet show the extra digit. It is also incompletely penetrant and an (apparent) unaffected person may have an affected child. This form of polydactyly is about 10 times more frequent in blacks than in Caucasians. From the study of various pedigrees, it is suggested that 2 phenotypically and possibly genetically different varieties exist. In type A, the extra digit is rather well formed and articulates with the fifth finger or an extra metacarpal. This type is inherited as an AD with marked penetrance. In type B (pedunculated postminimi), the extra digit is not well formed and is frequently in the form of a skin tag. The genetics of this type is more complicated. Using a 5 generation Indian family in which 15 individuals were affected a genome wide search showed linkage to markers on chromosome 7 and a candidate gene was identified. In a large Turkish family, the trait was linked to DNA markers on 13q. This was referred to as type A2 and the form in the Indian family as A1.
Phenylketonuria (PKU) is an AR trait. It is called an inborn error of metabolism because it is due to the malfunctioning of an enzyme. Classic PKU is due to mutations in the gene for phenylalanine hydroxylase (PAH) which converts phenylalanine to tyrosine. It is common enough (1/104 live white births) and serious enough (severe mental retardation) to be included in the newborn screening programs of all states. The lack of PAH causes toxic levels of the amino acid, phenylalanine, to build up in the blood which then damages the brain. A diet restricted in the amount of phenylalanine can prevent the serious mental retardation. The newborn screening is called the Guthrie test. A drop of blood from a baby who has had some food (milk) is dried on a small filter paper disk. The disk is placed on an agar plate containing B. subtilis and thienylalanine. If there are high levels of phenylalanine in the baby's blood, the growth of the bacteria will not be inhibited by the thienylalanine and you will observe bacterial growth around the test disk. A positive Guthrie test is followed by a quantitative assay of phenylalanine and tyrosine in the baby's blood before a definitive diagnosis of PKU is made. There is allelic heterogeneity due to different mutations in the phenylalanine hydroxylase (PAH) gene. Four common alleles account for the majority of cases among northern Europeans. However, there are other alleles in other ethnic groups. The frequency of the different alleles varies with different ethnic groups and many people affected with PKU are compound heterozygotes. Some of the mutations result in milder phenotypes. Mutations in genes at other loci can also cause hyperphenylalanemia. These genes are involved in the synthesis of tetrahydrobiopterin (BH4) or in its regeneration. BH4 is a cofactor in the reaction which converts phenylalanine to tyrosine which is carried out by PAH. One such gene is one that codes for the enzyme dihydropteridine reductase (DHPR) which reduces quininoid dihydrobioterin (qBH2) to tetrahydrobiopterin (BH4). Treatment is very different for these patients. Maternal PKU leads to severe MR in the fetus due to the high levels of phenylalanine in the blood. Therefore, females must remain on the restricted diet if they are to have normal children.
Tay Sachs Disease (TSD) is an AR inborn error of metabolism. It belongs to a family of genetic disorders known as the lysosomal storage diseases. Lysosome which are found in all cells of the body contain a variety of acid hydrolases which break down a variety of substrates which are large molecules. If any one of the hydrolytic enzymes is missing, there is a build up of the substrate in the lysosome. The organs affected depend on which enzyme is missing and where its substrate is primarily found. The enzyme missing in Tay Sachs disease is hexoseaminidase A, an enzyme that cleaves monosaccharides from oligosaccharides which are part of a lipid known as a ganglioside. These gangliosides are found primarily in the CNS and the lysosomes become engorged with these lipids.The accumulation kills the cells in the brain and CNS and the result is death in infancy. There is no known cure. Effective screening programs have been implemented among Ashkenazi Jewish communities where the carrier frequency is 1/27 as opposed to about 1/300 among other ethnic groups. Prenatal detection is also possible. The gene has a high incidence among some French Canadians as well. The higher incidence in some ethnic groups is due to founder effects. The higher incidence may be due to some as yet undiscovered heterozygote advantage. As in all cases we study, there is alleleic heterogeneity and locus heterogeneity. Some alleles which only reduce the enzyme activity result in a later onset.
Xeroderma pigmentosum (XP) is a disorder with locus heterogeneity. The defects are in the DNA excision repair pathway for UV damage. UV causes a TT dimer to form which must be identified and excised by an endonuclease, a section then removed by an exonuclease, a DNA polymerase and finally a ligase. This presents us with at least 4 required enzymes and each enzyme can be a multimer. Complementation tests have shown at least 9 complementation groups. The interpretation is that there are at least 9 genes involved in XP. Some forms affect the CNS and some do not. Skin cancer is initiated by UV mutations and progresses to cancer when these cells are not triggered by normal processes to go into apotosis.
Albinism exhibits locus and allelic heterogeneity. Albinism is often divided into tryrosinase positive and tyrosinase negative types. In oculocutaneous albinism known as OCA type IA (classic OCA) tyrosinase activity is absent. In type IB (yellow mutant) there is a partial deficiency of tyrosinase activity. There is some clustering of missense mutations in tyrosinase one such site is the binding site for copper, a cofactor for the enzyme. OCA type II individuals make a small amount of pigment and have light yellow hair. Both type I and type II lack visual pigmentation but type II has milder visual defects than type I. In type II, the major gene is referred to as the P gene which is thought to be involved in tyrosine transport into the cell. P refers to the pink-eyes of the phenotype. A temperature sensitive albinism has been reported in a few individuals who have white hair in warmer areas of the body and darker hair in the cooler extremities. This is the same as albinism in Siamese cats and Himalayan rabbits. Females heterozygous for an XR form of albinism show pigmented and non-pigmented patches in the in retina due to the mosaicism produced by Lyonization.
Lesch Nyhan syndrome is due to the deficiency of an XR enzyme, hypoxanthine phosphoribosyltransferase (HPRT). The extreme phenotype of self-mutilation, mental retardation, spasticity and high levels of uric acid leading to gout is associated with the lack of the enzyme. This enzyme is involved in purine metabolism which can affect levels of neurotransmitter among other things. There is allelic heterogeneity and other mutations in the gene lead to milder phenotypes such as gouty arthritis.
Fragile X is another X linked trait we have already studied as an example of a TNR disorder. Both males and females can be affected. Approximately 50% of the male offspring of a carrier mother and 30% of her daughters will be affected. Non manifesting carrier males can pass the premutation to their daughters who have a high risk for having affected offspring since there is high probability of expansion in premutation females than premutation males. The gene, FMR-1, is expressed in the CNS but its exact function is not known. The TNR (CGG repeats) occurs in the promoter region of the gene and when greatly expanded leads to methylation and inactivation of the gene.
Duchenne Muscular Dystrophy (DMD) is caused by mutations, mostly deletions, in the unusually large gene for a large rod-like protein called dystrophin. Dystrophin has some homology to spectrin, an actin-binding-protein and has been found in smooth, skeletal and cardiac muscle. It is localized to the cell membrane, confirming its role in the cytoskeleton. DMD is due to the complete absence of dystrophin usually caused by large deletions. Another disorder, Beckers Muscular Dystrophy (BMD) is also due to mutations in the dystrophin gene. However, BMD is milder than DMD because the mutations result in either less dystrophin or an abnormal dystrophin instead of no dystrophin.
Another XR disorder we have already discussed is androgen insensitivity/testicular feminization (AIS/Tfm). This disorder is both sex linked AND sex limited. It is sex linked because its gene, the androgen receptor, is on the X chromosome. It is sex-limited because it only manifests in XY fetuses. There are a variety of mutations in the androgen receptor (allelic heterogeneity) and there are also a wide variety of phenotypes produced by the different mutations in this gene. The phenotypes range from an phenotypically normal male who is merely infertile to a person with the outward appearance of a female but who has testes and no female internal plumbing.