BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Mutations
Mutations are any permanent heritable change in the genetic material which is passed on to daughter cells. They may or may not be revealed in phenotypic change. Mutations can be categorized many ways. They can occur in the genetic material of both somatic and germinal cells (nDNA) and also in mitochondrial DNA (mtDNA). They may involve whole chromosomes or parts of chromosomes leading to aneuploidy, structural aberrations, and microdeletions, all of which involve many genes. They may occur within exons, introns, splice sites (IVS8G-T,+1), and regulatory regions both 5' and 3' to genes. They may involve deletions or insertions or substitutions of one or a few bases. They may involve the insertion of many copies of a trinucleotide repeat. They may be point mutations involving one or a few bases. They may be silent, quiet, missense, or nonsense mutations (ter) depending on how they change the amnio acid sequence of the gene product. They may cause a frameshift in the reading of the messenger RNA so that the amino acid sequence is incorrect and the protein product is prematurely terminated by encountering a stop codon. Mutations can be caused by defects in DNA replication. DNA repair systems, or they may be the result of environmental mutagens. They often occur where there are repetitive sequences that cause mispairing of homologous regions with resulting duplications or deletions of whole genes or regions of genes (e.g. colorblindness, Duchenne Muscular Dystrophy, Congenital Adrenal Hyperplasia, alpha thalassemia). They may result in a product which is antigenically similar to the normal gene product (CRM+) or they may result in a null allele which is one that does not produce a functional product or does not produce product at all (CRM-).
Polymorphisms
Any permanent change in the genetic material can be referred to as a polymorphism if it has a population frequency of at least 1%. When speaking of changes within a single gene these differences may be referred to as alleles. For example, the A, B and O alleles are polymorphisms that result in the A, B, O, AB blood types. Some polymorphisms are neutral, some are bad for us and some maybe beneficial...if not immediately, maybe later on. Since mutations occur at any place within a gene, there are usually many different alleles for each gene. Some may be neutral and some may be deleterious. It is common in recessive disorders to find the patient is a compound heterozygote. This means that they have two defective genes but they are not mutated at the same site within the gene. Different mutations within the same gene is referred to as allelic heterogeneity. This presents a problem for population based genetic screening for disorders such as cystic fibrosis (CF) since it is possible only to test for the more common mutations for CF. Often, however, there are "common mutations" in the population for which people are tested. For example, there is a common mutation for beta thalassemia that was useful for population screening in Italy. Tay Sachs carriers can be picked up by enzyme assay but there are three common alleles which can be tested for by DNA analysis.
Mutations are the source of variety upon which evolution acts. Natural selection allows the most "fit" genotypes to survive to reproduce. In prokaryotes which are haploid, mutation is the primary source of variation since sexual recombination is rare. An example of selection which has profound effects for our health is antibiotic resistance of bacteria. The resistance is due to the selection of bacteria which have mutations that allow them to bypass the effects of the antibiotic. The antibiotic resistant bacteria survive to reproduce in that milieu. Their DNA can be taken up by unrelated bacteria through a process known as transfection. Thus the transfected bacteria are also antibiotic resistant. Diploid organisms can harbor mutations without expressing them immediately since they can have a normal allele to cover for them (in the heterozygous state). Gene duplication (often in tandem) and diploidy (two copies of each gene) allow us to acquire and retain mutations which are bad, neutral or good. The neutral or bad (at the time) could prove useful for the species in the future.
Biochemical individuality goes beyond SNPs (single nucleotide polymorphisms) and other mutations. It includes transcription control (enhancers, etc.), the interactions of allelic products, epigenetic gene controls (e.g., methylation patterns) which can persist over more than one generation, and other various networks of interactions of gene products.
Sporadic or new mutations
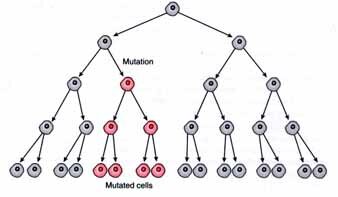
Mutations can occur in meiotic or mitotic cells. Germinal mutations are those changes that occur in the germ cells (gametes). They are be passed on to the next generation. New (sporadic) mutations that occur in the gonad may give rise to only one or a few gametes with the mutation depending on whether the mutation occurred in a mitotic feeder cell. Therefore, a germ line mutation can result in gonadal mosaicism and if the mutation shows a dominant pattern of inheritance, it can result one or more affected children. Obviously, mutations are often recessive and these will not show up immediately.
Genetic mosaics
If mutations occur post-zygotically, they produce a genetic mosaic. New mutations that arise in the early embryo produce an individual who is a phenotypic/genotypic mosaic. A variable number of cells will carry the mutation and the person may or may not be affected depending on the number and location of the cells which receive the mutation. Such a mutation may or may not be in the person's germ line and the mutation may be present in some germ cells but not all (gonadal mosaicism). If the mutation is in the germ line cells it will be transmitted to the offspring.
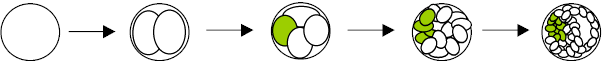
Depending on the stage of development in which the mutation takes place, a greater or lesser percent of the resulting cells will have the mutation. The mosaicism can be in the fetus and/or the placenta. Chorionic villus sampling (CVS) which is done between 10 and 12 gestational weeks to detect mutations including chromosome abnormalities often shows mosaicism, 1-2% of the time. The chorionic villi are extra embryonic tissue and part of the placenta. It is not under the stricter selective pressure exerted on the embryo proper. Therefore, mosaicism is more frequently seen in CVS than in amniotic fluid cells which derive solely from the embryo. Approximately 0.1% of amnios show mosaicism. If the mosaicism is confined to the placental tissue, it is referred to as confined placental mosaicism (CPM). However, if the CVS culture contains over 5% abnormal cells, a follow-up amniocentesis is indicated to rule out mosaicism in the fetus. It has been estimated that 5% of normal birth placenta have mosaicism.
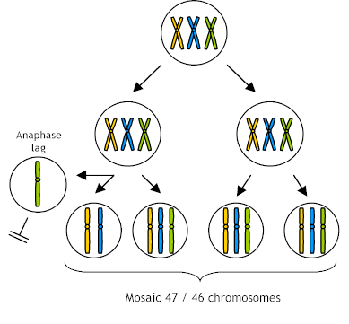
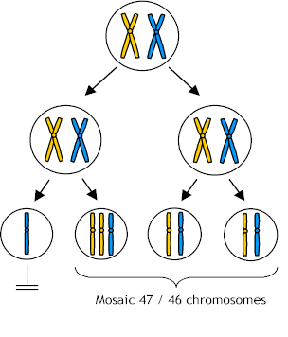
One (in)famous case of a CVS which showed trisomy 15 (three chromosomes 15) followed by an amniocentesis which showed the normal diploid number, resulted in a baby with Prader Willi Syndrome. The baby had only two chromosome 15's but they both came from the mother. This is referred to as uniparental disomy (UPD) which is discussed in detail in other lectures. There are genes on chromosome 15 which are inactivated in oögenesis, therefore, to be normal you must have the paternal gene from chromosome 15. This baby started out as a trisomy 15 and during embryonic mitosis, one of the 15's was lost in those cells that formed the embryo but the placenta retained the trisomic state. A trisomy 15 fetus would have been aborted while a trisomic placenta is tolerated.
Chromosome changes are also mutations. The chromosome anomalies which we have studied previously are called "chromosome mutations" to distinguish them from those that involve base changes or small amounts of DNA. Obviously there is a fine dividing line since there is a wide spectrum of changes ranging from the chromosome aneuploidies and gross structural changes to the microdeletions. All of these involve many genes. There are also normal chromosome polymorphisms involving the heterochromatin on chromosome 1, the satellites on the D and G group chromosomes, and the variable amount of heterochromatin on the q arm of the Y chromosome. Although these are examples of neutral chromosome polymorphisms, the other numerical and structural changes in chromosomes usually involve the addition or loss of chromosome material and they often have harmful consequences.
Point Mutations
Much of the time geneticists talk about mutations they are referring to "point mutations" which are changes in one or a limited few nucleotides within a single gene. Point mutations are changes in the DNA sequence involving one (or a few) nucleotides. They can occur anywhere in the DNA--coding or non coding regions. They produce polymorphisms in non coding regions which are useful in DNA fingerprinting. They can occur in tRNA and rRNA genes as well as in genes coding for polypeptides and they can occur in non coding control regions of genes. The possible effects of these mutations within exons and introns will be described. Many mutations only produce the normal "polymorphisms" found within a population. Everyone you know has some differences in their DNA sequences from you. For a genetic difference to be termed a polymorphism, at least 1% of the population must have the alternative form of the allele. Normal variation from which evolution selects is due to mutations. Changes in repetitive elements such as the STRs produce the wide variety of alleles used in DNA fingerprinting for forensic testing, paternity testing, identity testing and gene mapping.
In order to understand mutations and their effects, you must understand protein structure. Specifically, you must understand what kind of amnio acid changes in the primary protein structure will make differences. Some amino acids are similar to one another in their functional groups and others are very different. The substitution of a proline, for example, will put a bend into an alpha helix (secondary structure) while the substitution of alanine for glycine may not make a big difference in the conformation and function of a protein. Changes in the tertiary and quaternary structure of proteins will usually make a difference in the function. Obviously a change in the active site of a protein will change, at least, its effectiveness. Changes in allosteric sites or sites where cofactors are bound will also affect function. While changes can be for the better, most frequently they will not. Many mutations take place in non-coding regions and will have an effect only if the non-coding region plays a role in transcription or post-transcriptional processing.
Point mutations refer to those changes in DNA which occur within a single gene and includes mutations in the control regions and introns as well as exons. Point mutations can be divided into deletions/insertions which cause frameshift mutations and substitution mutations where one nucleotide is substituted for another. Deletions and insertions are called frameshift mutations because they throw the reading frame (translation) off. They result in truncated proteins because a termination codon is encountered in the transcript due to the frameshift.
Single nucleotide polymorphisms (SNPs) are identified in increasing numbers due to the relative ease of DNA sequencing. Clinical molecular laboratories detect these SNPs in the course of testing patient specimens. The clinical interpretation and significance of these sequence variations may not always be known with certainty. While some of these variations may be the cause of the disease, other most certainly are not. An example of a sequence variation which would not be the cause of a disease is one which does not produce an amnio acid substitution and which is unlikely to produce a cryptic splice site.
Types of point mutations

A. Deletions and insertions of nucleotides
Deletions or insertions, unless they are multiples of three, will cause a shift in the mRNA reading frame. So this type of mutation is usually called a "frameshift" mutation. These mutations usually result in encountering a premature termination codon during translation and, therefore, a shortened (nonfunctional) truncated protein. NF1 is an example of a disorder which often has truncated protein gene product. Even if no termination codon is encountered, the amino acid sequence will not resemble the non mutant sequence!
If three nucleotides are deleted the amino acid will not be in the final polypeptide but the reading frame will not be disturbed before or beyond that point. The most common mutation causing Cystic Fibrosis is F508 that deletes phenylalanine, amino acid 508, in the normal protein.
B. Substitutions of one nucleotide for another
Substitution mutations are those where one nucleotide is replaced by another nucleotide. They are called transition if a purine is replaced by another purine, a pyrimidine is replaced by another pyrimidine or a transversion if a purine is replaced by a pyrimidine or pyrimidine by a purine. Substitution mutations are responsible for the normal polymorphisms and for genetic disorders. They are also the ones responsible for evolution. Base substitutions can have one of several results in the gene product.
1. No change in the amino acid (sequence) = Silent mutations. This is due to the redundancy of the genetic code. More than one codon codes for the same amnio acid.
2. Change in the amino acid (sequence) = Missense mutations. If the substituted amnio acid has properties similar to the original amino acid this may have little or no effect on the protein function = quiet mutation However, if the substituted amino acid has properties that are different from the original amino acid, the result can be a nonfunctional protein, a less functional protein (leaky), or even a better functioning protein (rare). These mutations obviously cause changes in the 1o structure of the protein and can cause changes in 1. Overall conformation (2o , 3o , 4o ) with premature degradation of the protein. 2. Changes in the binding site (Km). 3. Changes in the catalytic site. 4. Changes in the allosteric site
3. Change from an amino acid codon to a termination codon = Nonsense mutations. This type of mutation shortens the length of the polypeptide chain. The effect on function depends on where in the sequence it occurs.
4. Change from a termination codon to an amino acid codon. This type of mutation lengthens the polypeptide chain. Translation will continue until another termination codon is encountered in the mRNA.
5. Change in the splice site consensus sequence. This type of mutation can occur at the acceptor (GT) or donor (AG) site, it can be the consequence of the formation of a cryptic splice site (a new acceptor or donor site) where there was none before or it can affect the binding of the proteins which are involved in splicing. Any of these mutations changes the pattern of RNA splicing (post transcriptional processing) and leads to exon skipping or insertion of some intron sequence into the mRNA. A mutation in another location within the gene that results in a "cryptic" splice site causes a mistake when it is interpreted as the "real" splice site.

Single Base Change Consequences
Repetitive Sequences: DNA Identity Testing and Role in Mutations
As mentioned earlier, VNTRs (variable number of tandem repeats) and STRs (short tandem repeats) are terms used for segments of DNA consisting of a multiply repeated short sequence element. The number of nucleotides in a repeat as well as the number of repeats at a given locus varies. These polymorphisms are clustered at many sites in the genome in non coding areas including introns. The number of repeats at homologous loci is variable and these polymorphisms are a major source of genetic markers for the Human Genome project, for forensics and for paternity testing (also for evolutionary relationships, degree of relatedness in breeding animals, etc.) These repetitive sequences are also called mini satellites (VNTRs) and micro satellites (STRs).
An example of a VNTR repeat is: GAATCGAATCGAATCGAATCGAATCGAATC. This sequence is made up of six tandem repeats of a pentanucleotide sequence, GAATC. Clusters of such repeats are scattered at many sites on all chromosomes. The number of repeats at each site is variable, ranging from 2 to more than 100. Each variant is an allele. Because there are many different VNTR loci and because many loci have dozens of alleles, heterozygosity is common. As a result, each person (except MZ twins) has a unique pattern of bands produced when many VNTR sequences are cut with restriction enzymes, run on gels and probed with the specific sequence. This pattern of DNA fragments is referred to as a DNA fingerprint. Sometimes you may see DNA probes used to identify polymorphic loci on chromosomes named thusly: "DXS52" which refers to a piece of DNA of the X chromosome, Single copy, #52 or "D16Z44" which refers to a piece of DNA of chromosome 16, Z means repetitive, #44 refers to some numbering system for the various pieces of DNA. Sometimes DNA probes are for a particular gene like Apo B and are named for the gene they probe.
Prior to the finding of these tandem repeats, the only polymorphisms available were the ABO blood groups and the HLA (human leukocyte antigens) loci (genes) on chromosome 6. These loci are extremely variable (polymorphic) and most individuals are unique for their haplotypes. Haplotype refers to the genotype of a group of alleles from two or more closely linked loci on one chromosome, usually inherited as a unit. Today some of you heard a lecture on the genetics of diabetes which pointed out that Type I diabetes is seen in association with specific HLA loci.
Repetitive sequences are often found to be involved in deletions and insertions of segments of DNA. The reason is that two homologous chromosomes more frequently misalign during recombination in the regions with the repetitive sequences, then when cross over occurs a deletion or insertion results. It has been shown that there are several microdeletion syndromes which have flanking repetitive sequences on either side of the deletion area. Insertions also occur but may not be noticed because the result is a milder phenotype.
In addition to point mutations there are duplications and deletions of pieces of DNA which are caused by repetitive elements in the genome. These repetitive regions can be pseudogenes such as the CYPB gene and the functional gene CYPA or the family of globin genes with similar sequences. In addition there are repetitive regions (non-coding) which flank the regions involved in microdeletions. These repetitive elements can result in a mismatching of the homologous regions (of chromatids). A subsequent crossing over between the mismatched regions results in a duplication of the intervening region on one strand and a deletion of the same region on the other chromatid. Repetitive sequences ranging from two to a few hundred nucleotides in length are found scattered over the entire genome.
Hemophilia A inversion mutation due to recombination between L1 repetitive sequences within an outside the F8 gene
Hemophilia A is due to a mutation in the Factor VIII (F8) gene. In a few patients with Hemophilia A, L1 (see lecture on the genome which describes LINES) sequences several kb long were found to be inserted into an exon in the factor VIII gene, interrupting the coding sequence and inactivating the gene. An inversion occurs through recombination between an L1 sequence located within intron 22 with one of usually two additional copies of homologous sequence that are located far from the F8 gene near the telomere of the long arm of the X chromosome. These telomeric sequences are in the opposite orientation relative to the intron 22 sequence.
Common Mutations Found in Genetic Disorders or Conditions
Hemophilia B: A single base change in the promoter of the factor IX gene results in a lower level of transcription and therefore less of factor IX.
Neurofibromatosis 1: Single base changes in the NF1 (tumor suppressor, neurofibromin) gene results in a premature stop codon (nonsense) in place of an amino acid codon and therefore truncated protein product.
Tay Sachs Disease: A single base change in hexoseaminidase A at the 5' end of the intron splice site causes the intron to not be removed and, therefore, there is a lack of a normal transcript and protein product. Another mutation is an insertion of four bases in the Hex A gene which caused a frameshift mutation, an early stop codon, and a truncated protein.
ABO Blood Group: A single base deletion in the glycosyl transferase gene is responsible for the A allele of the ABO blood system. It leads to a frameshift and altered reading frame.
Duplications and deletions of entire gene sequences are often due to mispairing of homologous regions of similar sequences that are near one another. An example of this is found in congenital adrenal hyperplasia (CAH). The responsible gene (CYP21B) on chromosome 6 in the middle of the HLA genes. There is a CAH pseudogene (CYP21A) nearby which can misalign with the "real" CAH gene and through unequal crossing over there is the loss of the good allele. The globin genes can also misalign due to their sequence similarity thereby causing a variety of mutations. Alpha thalassemia is due to the deletion of the alpha genes. There are two identical alpha globin alleles which can mispair. Hb Lepore which is part delta and part beta globin is due to (mis)alignment of the beta and delta globin genes. In Duchenne and Becker Muscular dystrophies, the gene is extremely large (largest gene found so far) and contains within it many repetitive gene sequences for the same repetitive domain found in the dystrophin protein product. The homologous chromosomes can misalign and crossing over will cause deletions and additions within the gene The presence of Alu sequences (the repetitive SINES discussed earlier in the course) can also cause misalignment and unequal crossing over.
Since genes are several kb long, many different kinds of mutations occur within the exons or introns. Mutations can also occur in the promoter site, the 5' UTR, the 3' UTR, or in a transcriptional control region. The existence of many mutations within a gene is called "allelic heterogeneity." Each person with a disorder may harbor different mutations. A person with an autosomal recessive (AR) disorder often has two different mutant alleles. If this is known, the person is referred to as a compound heterozygote. This is often the case in cystic fibrosis.
Repetitive sequences abound in the genome. Some are large and some small but they are often responsible for mispairing and consequently cause deletions and additions. The trinucleotide repeat disorders (TNR) are due to mutations involving repeated nucleotide sequences of three bases. The TNRs interfere with the normal functioning usually of a single gene. Other repeat sequences such as VNTRs (variable number of tandem repeats of 7 or more bp or mini satellites) and STRs (short tandem repeats of 2 to 4 bp or micro satellites) show variation in the number of repeats and are very polymorphic in all populations. These VNTRs and STRs are found in the non coding sequences of the DNA including introns. They do not usually cause a problem and are extremely useful in gene mapping and DNA profiling.
While mutations are permanent changes, there are also non permanent changes in the genetic material. These are referred to collectively as epigenetic changes. Included are temporary or reversible changes such as the inactivation of X chromosomes and the silencing of specific genes during spermatogenesis or oögenesis. The latter changes result in parent of origin effects. They are reversed when the chromosome or genes pass through offspring of the opposite sex. At one time it was thought that the genetic contribution of both parents was equal but now we know that is not entirely true. Anyone who knows about hinnies and mules could have told us this a long time ago. A hinny is a hybrid of a donkey mother and horse father and is quite different from a mule which is the product of a horse mother and donkey father. (As we will learn later, parent of origin effects also include more permanent genetic changes such as the greater expansion of trinucleotide repeats mutations in one or the other parent.)
The Main Classes of Mutation
|
Deletions
|
1 bp to megabases |
| Insertions | Includes duplications |
| Single base substitutions | Missense mutations replace one amino acid with another in the product Nonsense mutations replace an amino acid codon with a stop codon Splice site mutations create or destroy signals for exon-intron splicing |
| Frameshifts | Can be produced by deletions, insertions or splicing errors |
| Dynamic mutations | Tandem repeats that often change size on transmission to children |
Nomenclature for describing mutations:
Amino acid substitutions. Use the one letter codes A=ala, C=cys, D=asp, E=glu, F=phe, G=gly, H=his, I=iso, K=lys, M=met, N=asp, P=pro, Q=gln, R=arg, S=ser, T=thr, V=val, W=trp, Y=tyr, X=stop. R117H or Arg117His - replace arginine 117 by histidine (the initiator methionine is codon 1). G542X or Gly542Stop - glycine 542 replaced by a stop codon.
Nucleotide substitution. The A of the initiator ATG codon is +1; the immediately preceding base is -1. There is no zero. Give the nucleotide number followed by the change. If necessary use g. and c. to designate genomic and cDNA sequences. For changes within introns, when only the cDNA sequence is known in full, specify the intron number by IVSn or the number of the nearest exon position. 1162G>A -replace guanine at position 1162 by adenine. 621 + 1G>T or IVS4 + 1G>T - replace G by T at the first base of intron 4; exon 4 ends at nt 621.
Deletions and insertions. Use del for deletions and ins for insertions. As above, for DNA changes the nucleotide position or interval comes first, for amino acid changes the amino acid symbol comes first. F508del - delete phenylalanine 508. 6232-6236del or 6232-6236delATAAG - delete 5 nucleotides (which can be specified) starting with nt 6232. 409-410insC - insert C between nt 409 and 410.
Null alleles are those that do not produce a protein product and are "CRM- or CRM negative." CRM + or CRM positive mutant produce a protein product that resembles the wild-type enough that they cross react with antibodies to the wild-type protein. CRM, therefore, stands for "cross reacting material." A person with a CRM+ mutation may be able to be treated by providing them with more substrate, cofactors or coenzymes that work with the normal enzyme or other protein product. Also, they will not make antibodies to the enzyme if enzyme therapy is possible as in some of the lysosomal storage diseases. Those with a CRM- mutation, in time, will make antibodies to any enzyme administered since to them it is a "foreign" protein.
A null allele which results in the loss of activity of a gene is sometimes better than making a defective protein which interferes with a normal cellular process. A null allele of the pro alpha collagen gene causes a milder form of osteogenesis imperfecta (OI) than the mutations which produce abnormal forms of the protein and the more severe forms of osteogenesis imperfecta. The TNR disorders are also disorders where the mutation causes the accumulation of the abnormal gene product which interferes with the normal gene product. Another example are the fibroblast growth factor receptor disorders, which include achondroplasia and other chondrodystrophies, are due to mutations which prevent the proper dimerization of the fibroblast growth factor receptor. If the abnormal monomer associates with the normal one, it is not functional.
Allelic Heterogeneity
Allelic heterogeneity refers to the presence of many mutant alleles of the same gene present in the gene pool. Allelic heterogeneity arises because genes are very long, several kb, and mutations can occur in any site within the gene and its control regions. Infantile, juvenile and/or adult onset forms of the same genetic disorder can be due to allelic heterogeneity. In these different forms, the patients have combinations of alleles some of which make at least some of the gene product (leaky) and others make less or none. For example, two null alleles or alleles that do not make enough of an enzyme will result in an earlier onset than one "leaky" allele and one null allele. Two "leaky" alleles may allow a person to not show symptoms until adulthood. A good example of this is with Tay Sachs Disease, a lysosomal storage disease. The three most common Jewish mutations found in over 97% of obligate Jewish carriers are: base substitution (gly269ser), insertion (+TATC) and a splice junction mutation (1IVS12). We have a student at CSUDH that has the gly269ser mutation and the insertion (+TATC). He is a compound heterozygote. He received the base substitution allele from his mother and the null allele from his father. His hexoseaminidase A enzyme level was 4.7% which is indicative of the late onset disease. The insertion or splice junction mutations are the ones found in infantile Tay Sachs and are null alleles. People with the later onset will have the base substitution mutation (called the adult allele) and one of the two null alleles found in the infantile form. It is amazing that 4.7% of the enzyme activity can maintain life. Carriers of either of the infantile forms will make 50% (gene dosage effect) and are perfectly healthy.

Pictorial Representation of some of the known mutations in the X linked Androgen Receptor Gene
Allelic heterogeneity is a problem, for example, in cystic fibrosis (CF) testing. CF is an AR condition with a carrier frequency of about 1/25 in the European Caucasian population. The most common mutation is the deletion of 3 bases in the CFTR (CF trans membrane conductance regulator) gene which eliminates a phenylalanine residue at position 508. The mutation is called F508 with meaning deletion, F meaning phenylalanine, and 508 referring to the amino acid position within the gene product. This mutation accounts for 70-80% of mutant alleles among the northern European Caucasian population which can be attributed to a "Founder effect." However, it is less common in other populations. There are at least 70 other "common" mutations. Therefore, at the present time, carrier screening is still considered controversial by some and prenatal diagnosis has been confined to "at risk" pregnancies as defined by the previous birth of an affected child.

Incidentally, allelic heterogeneity can result in disorders that do not have the same phenotype in the CFTR gene and many others such as the various hemoglobinopathies due to mutations at different sites within the beta globin gene.
Locus heterogeneity
Locus heterogeneity refers to the fact that clinically similar disorders may be due to mutations in different genes at a variety of loci. Xeroderma pigmentosum and the mucopolysaccharidoses are good examples of locus heterogeneity. It can be shown that different loci are involved by performing complementation tests. One grows cells taken from affected individuals together in somatic cell culture. If the defect is corrected by the two cell lines growing together then it is assumed they have mutations in different genes. For example, let us say that genes A and B each code for separate proteins necessary for a particular biochemical pathway (e.g., gene A codes for the specific endonuclease that identifies TT dimers and gene B codes for the exonuclease which digests away the loose ends produced by the endonuclease). When the cells from person A and B are cultured together, the normal gene product from gene B is made by the person with a mutation in gene A and the normal gene product from gene A is made by the person with a mutation in gene B and the normal functioning of the pathway is restored. This is referred to as "complementation" which was first shown in viruses. So the number of complementation groups corresponds (roughly) to the number of proteins required for the specific metabolic process. There are at least 5 complementation groups for XP. Other clues to locus heterogeneity are differences in the clinical features (e.g., severity, age of onset), chance matings between affected individuals that result in normal offspring (e.g., different types of AR deafness), linkage analysis shows the genes are not the same, or CRM status.
PKU is an disease which has both allelic and locus heterogeneity. High levels of the amino acid phenylalanine, hyperphenylalaninemia, is usually due to mutations in the gene for phenylalanine hydroxylases (PAH) but can also be due to mutations in the metabolic pathways which make the cofactor BH4 or which regenerate it. BH4 is a necessary cofactor for phenylalanine hydroxylase functioning properly. This is an example of locus heterogeneity where mutations in different genes (at different loci) cause the same clinical phenotype (genocopies). Allelic heterogeneity also exists for PKU. There are 4 common mutations in PAH which cause PKU. Three are amino acid substitutions, only one of which causes classical PKU, and the fourth is the most common PKU mutation which is in the exon 12 donor splice site where GT is mutated to AT. This causes skipping of the 12th exon during RNA splicing and a premature termination codon in the new sequence generates a truncated and unstable enzyme. In most populations the majority of patients with PKU are compound heterozygotes (i.e., have two different abnormal alleles). This is reflected in clinical observations of phenotypic heterogeneity. Since the screening tests measure only the amount of excess phenylalanemia and do not look at the DNA, the condition can be detected no matter what the etiology.
Many disorders show either allelic or locus heterogeneity. Sickle cell anemia is an exception since the mutation is a single base change (and a single amino acid change) detectable by DNA analysis. Although even here we have HbC which is a mutation in the same codon and which causes a milder form of the disease. There are many compound heterozygotes with HbS HbC. Due to allelic heterogeneity, many people with AR genetic disorders such as CF and Friedreich ataxia are compound heterozygotes.
The terms dominant and recessive refer to the pattern of inheritance of a disorder but reveal something about the gene function. A dominantly inherited disorder generally means the mutant allele is producing a product which changes or interferes with a normal process. These traits are said to be due to gain of function, gain of malfunction or dominant negative mutations....as the case may be. They can also be due to haplo insufficiency when the expression of both alleles is required for normalcy. A recessively inherited disorder/trait would conversely be seen as a loss of function. A recessive trait is one in which both alleles have to be non functional before the trait or disorder manifests. Another way of looking at it is that one wild-type allele is sufficient to produce enough gene product to cover for a mutant allele.
However, some dominant traits, such as NF1 and retinoblastoma, are due to the inheritance of one (null) mutant allele and one good allele. But the activity of the good gene which you also initially inherited, is knocked out by a somatic mutation resulting in the loss of the heterozygosity (LOH) you enjoyed until then. Codominant inheritance merely means that you can detect the presence of both alleles by their products or activities. This is possible at some level for all genes but we generally mean at the easily observable phenotypic level. Haplo insufficiency is a term given to a disorder due to the loss of activity of one allele. While 50% of the gene product may not be a problem for some cellular processes, this is not always true. This is the situation in the disorder called familial hypercholesterolemia (FH). In FH there are insufficient low density lipoprotein (LDL) receptors in the heterozygote to prevent high levels of cholesterol from building up in the blood vessels. Homozygotes have extremely early onset and die in their teens. So FH is said to be dominantly inherited but a double dose in a homozygote is an early lethal.
There are differences in the origin, rate and types of mutations arising in the male and female. The germ line mutation rate in human males, especially older males, is generally much higher than in females, mainly because in males there are many more germ cell divisions. There are some exceptions. NF1 (AD) and Duchenne Muscular Dystrophy (X-linked) are single gene traits that show only a slight paternal bias and a small paternal age effect. These genes are extremely long. DMD has 55 exons and NF1 has 59. Their mutations rates are extremely high. Many of the mutations in large genes are intragenic deletions. Such large deletions are not tolerated in smaller genes since the results would be genetic lethals. It is hypothesized that base substitutions occur primarily in males and are age dependent and small chromosomal changes (mainly intragenic deletions) are not age dependent because they occur by different mechanisms. The rate of deletions is higher in females than in males. For DMD, 93% of point mutations were from sperm, and 87% of deletions were maternal in origin. Aneuploidy (trisomy or monosomy) is the most commonly identified chromosome abnormality in humans. It occurs in at least 5% of all clinically recognized pregnancies. Of course, it has long been known that chromosome aneuploidies, with the exception of XXY, XYY, XO are associated with increasing maternal age....93% of informative cases were of maternal origin. The incidence of aneuploidy in sperm is believed to be 1-2% and in oocytes, approximately 20%.
Cancer
Cancer is a genetic disease but only some cancers are "familial." There are familial cancers and sporadic ("environmental") cancers. Familial cancers are ones which occur more frequently in first degree relatives since it is more likely that they will have also inherited the predisposing mutation. Inherited and non inherited cancer are both due to mutations in genes that control the cell cycle and DNA replication and repair.
Cancer is clonal; it originates with a single cell which because of the loss of control, continues to divide to form the tumor or cancer. Genetic and biochemical analyses have confirmed that all cancer cells in an individual have arisen from a single original precursor cell. It is assumed that the original cancer cell was transformed when it underwent a mutational event (or a series of mutations) which resulted in the loss of control of cell proliferation. That cell has an advantage over its normal neighbors because its growth is unregulated (it is an example of "evolutionary" selection).
Mutagens and carcinogens (cancer producing agents) are overlapping terms. When a mutation occurs in a somatic cell (somatic mutation), it may result in cancer or tumors if it occurs in a tumor suppresser gene, DNA repair genes or cell cycle control genes. Somatic or post zygotic mutational events are involved in all cancers. Somatic cell gene mutations or chromosome rearrangements can cause neoplasia (cancer). Mutations can often be characterized: melanomas are often initially UV induced (CG to TA and CCGG to TTAA), lung cancers show smoke induced mutations, and hepatocelluar cancers reveal aflatoxin induced mutations.
The genes involved in cancer are referred to as proto-oncogenes or tumor suppressor genes. Tumor suppressor genes and proto-oncogenes are normal genes normally involved in the control of cell growth. When mutated, however, the result is loss of control of cell division, apoptosis, and differentiation. In general, one can think of cancer as being due to mutations in proto-oncogenes or tumor suppressor genes or in genes which predispose one to mutations (e.g., lack of DNA repair). Proto-oncogenes which are normal genes involved in cell growth become oncogenes by mutation. A single mutation can cause the gene to be permanently on and no longer subject to normal cellular controls. A virus can carry an oncogene (e.g., retroviruses), and oncogenes can be activated by gene amplification (some neuroblastomas and breast cancer) or chromosome rearrangements (e.g., Burkitt lymphoma). When a proto-oncogene is mutated (or over expressed) the mutation causes the production of an inappropriate protein which is not responsive to the normal cellular controls. Therefore, the single mutation acts in a dominant fashion. Reactivation of an embryonic gene involved in cell proliferation or the production of a mutant protein which is no longer responsive to cell controls leads to uncontrolled cell proliferation. This situation can result from a point mutation in an protein which is part of an important signal transduction pathway, mutations in a receptor protein which is consequently always "on," or rearrangements of control elements of genes causing a gene product to be made constitutively. The majority of cancer gene mutations that have been identified have been found to be in protein kinases which are involved in cell transduction pathways.
Inherited cancer occurs at an earlier age than sporadic cancer and is often bilateral if there are two organs (eyes, breasts, kidneys). Incidentally, men can pass breast cancer genes to their offspring and men can also get breast cancer. Inherited cancer is due to the inheritance of a predisposing germ line mutation thus only a single somatic mutation is necessary to eliminate the normal gene function. For example, in inherited childhood retinoblastoma, a childhood eye cancer, the child inherits one mutation in a tumor suppressor gene and the other normal allele is mutated or lost later (post-zygotically). Inherited breast cancer is a similar story. You inherit one mutation in a tumor suppressor gene from a parent and the second mutation occurs due to loss of heterozygosity at that locus.
Since two mutations are usually needed to knock out the normal gene function, these tumor suppressor genes are said to act recessively. This can be explained by the Knudson (two hit) hypothesis. The Knudson hypothesis proposes that the germ line event in familial retinoblastoma is inactivation of one allele of an autosomal tumor suppressor gene. The carrier would be heterozygous for the mutant allele but the normal allele product (a tumor suppressor) would be produced in sufficient quantity to prevent the growth of a tumor. However, a second mutational event which occurs in the normal allele in a somatic cell of the carrier would result in the loss of heterozygosity. The loss can be caused by deletion, somatic crossing over, chromosome loss, etc. with the subsequent loss of the tumor suppressor protein. This state of affairs would lead to unregulated growth and the result would be a tumor.
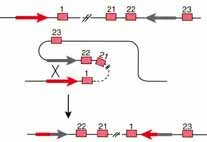
It can be somewhat confusing at first to learn that tumor suppressor genes are inherited in an autosomal dominant pattern (AD) but the cancer is due to loss of heterozygosity which is what happens in autosomal recessive (AR) disorders. The point, however, is to remember that the normal function of the genes we call tumor suppressors is to put breaks on cell proliferation and one functional gene is sufficient to carry out the duties of controlling cell proliferation. The confusion is that we named them for the abnormal function which operates in a recessive manner. Loss of heterozygosity can occur by somatic mutation, gene deletion, loss of a chromosome during mitosis, silencing of the normal gene or recombination between homologs during mitosis (despite what you have been told previously, recombination between homologs can and does occur during mitosis...another example of "whatever can happen, does happen").
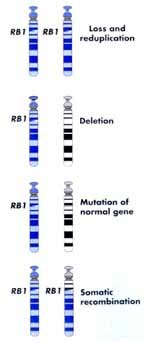
Mechanisms involved in the Loss of Heterozygosity (LOH)
Neurofibromatosis 1 is an autosomal dominant disorder caused by mutations in a tumor suppresser gene located on 17q11.2. The gene is very long, 350 kb with 59 exons and very apt to accumulate a mutation both in gonadal and somatic cells. The gene is expressed in a wide variety of human tissues and tumors result when there is a loss of heterozygosity (LOH) due to mutations (or other events) in somatic cells which disable the "good" tumor suppresser allele. This mechanism is also operative in retinoblastoma which was mentioned earlier. In NF1 the loss of the normal allele function causes an impaired regulation of the GTP/RAS pathway.
As many as fifty percent of NF 1 cases are due to new mutations, usually of paternal origin. The mutation rate of 1/10000 gametes is one of the highest for a human disorder and is due to the size of the gene. Although the gene has been mapped, since many different mutations have been found, diagnosis by direct DNA analysis is not possible. Linkage studies in informative families can be done if DNA markers close to the NF1 gene in the parents are "informative"....different enough to be identifiable. Gonadal mosaicism has been demonstrated in some unaffected fathers of children with NF1. Patients of affected mothers have a more severe disease. (Duchenne Muscular Dystrophy is an even larger gene, 2.3 MB with 79 exons, with the same mutation rate. One third are due to new mutations.)
The gene product is a 250 kD protein called neurofibromin which is most abundant in the nervous system. Eighty percent of inherited and sporadic cases encode a truncated protein due to premature protein termination during translation. [Deletions or additions of a non multiple of three bases, are called frameshift mutations and result in termination codons within the transcript where they should not be.] Diagnostic molecular testing for NF1 involves testing for the resulting truncated protein. The protein truncation assay uses 10 ml of whole blood (EDTA purple top tube). $500 for the first person and $350 for other family members. PCR primers with appropriate sequences for transcription and translation amplify five overlapping segments of the coding region. Polypeptides are synthesized in vitro from RT-PCR products. The wild type (normal) allele makes a 75.8 kD peptide, the mutant alleles make a shorter peptide.
(The following is the way I explained NF1 to a person who e-mailed her questions from Malaysia. It is redundant but may help.)
"Think about it this way....each gene you have produces a product. A mutant gene either produces a mutant product or none at all. Since we have pairs of genes...one from each parent....those who have NF1 have inherited one good gene and one mutant gene. In the case of NF1, the person has one good gene which makes a tumor suppressor protein but the other gene of the pair does not make a tumor suppressor. Everything is fine in those cells which have the one good functional gene. However, it is inevitable that some cells of the body will experience a mutation in the "good" NF1 gene (these mutations are called "somatic" mutations to distinguish them from "germinal" mutations which are in the egg or sperm). When this happens, that cell will not be able to make ANY tumor suppressor protein at all and so the cell goes out of control and divides and develops into a neurofibroma or glioma. A baby born with NF1 has not lived long enough to accumulate these somatic mutations but sooner or later it will happen. At what age the somatic mutations begin to occur is a matter of chance or exposure to mutagens. Similarly, the number of fibromas depends on the number of somatic mutations that occur in the patient. The Lisch nodules and optic gliomas are also "fibromas" and the cafe au lait spots are just another manifestation of how this gene mutation is expressed. There are families which have inherited cafe-au-lait spots with no fibromas so cafe au lait spots can be due to entirely different genes. When different genes produce similar phenotypes this is referred to as genetic heterogeneity. When a single gene produces seemingly unrelated problems this is known as pleiotropy. When a single disorder shows variation within a family, all of whom share the same mutation, this is called variable expressivity. No two people with NF1 will manifest the disorder in exactly the same way so there is variation in expression. However, everyone with the mutation will show some symptoms and so the gene is said to be fully penetrant. NF1 is inherited in an autosomal dominant pattern but about half of those affected are due to "new mutations." This means that they do not have an affected parent but that one of the parents gave them a newly mutated gene in the egg or sperm."
