BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Clinical Genetics
Human Blood Groups
ABO
There are many human blood groups and several are polymorphic. The blood groups are defined by the antigens found on the surface of red blood cells (and sometimes other cells as well). The ABO system is the best known because of its importance in transfusions. The locus is on chromosome 9 and there are three alleles (the first use of the term multiple alleles). The gene codes for glycosyl transferases which add sugars to complex oligosaccharides attached to integral membrane proteins and membrane sphingolipids. The antigenic specificity is due to the terminal sugars. An allele at another locus produces the H antigen which is the "O" antigen. If the H allele is mutated, no H antigen is produced. The absence of the H antigen is referred to as the Bombay phenotype, it is an AR condition. Such people have ABO alleles but they are not expressed, the standard blood typing picks them up as "O" even though their genotype for the ABO locus may not be OO. The H antigen is modified by the action of glycosyl transferases coded for by the A and B alleles. The O allele does not modify the H antigen because of a frameshift mutation in the gene. The A allele glycosyl transferase adds N-acetylgalactosamine to the H antigen and the B allele glycosyl transferase adds D-galactose to the H antigen. The A and B alleles act co dominantly, both producing their gene product. The O allele is recessive to both A and B since it does not modify the H antigen. There are 6 genotypes (AO, AA, BO, BB, AB, OO) in the population and four phenotypes (A, B, O, and AB).
The ABO blood group types differ from other blood groups in that we have pre formed antibodies to the A and B antigens (unless we are AB blood type). This happens even though we have not been exposed to the A or B antigens. Type A individuals made antibodies to the B antigen; type B make antibodies to the A antigen, and type O make antibodies to both the A and B antigens. AB makes no antibodies to either A or B antigen. Therefore, care must be taken in transfusions. In general, it is best to give blood of the same type as the person transfused. However, since type O has no antigens, it can be given to all the people with the other types. The antibodies made by the type O person are of no or small consequence since they will be diluted out.
The Rh Blood Types
The second most clinically important blood group is the Rh (rhesus, named after the monkey) blood group. The locus is on chromosome 1. This system is genetically complex and we will not go into the details (you may do so on your own, if you care to). In general, people are either Rh+ or Rh- meaning they either have the Rh antigen on the surface of their rbcs or they don't. For convenience, we can say there are two alleles and that the Rh+ allele which makes the antigen, is dominant to the Rh- allele which does not. An Rh+ person (phenotype) can be homozygous or heterozygous.
There are no pre formed antibodies such as are found for ABO. A problem can arise when an Rh- person receives a transfusion or Rh+ blood or when an Rh- mother has an Rh + child. The Rh+ antigens introduced into the individual stimulate the production of antibodies to the Rh antigen. The person is then said to be sensitized. A second transfusion with Rh+ blood can produce anaphylactic shock and death.The first pregnancy of an Rh- mother with an Rh+ fetus is usually without a problems (unless the mother was sensitized earlier) because the baby's erythroblasts do not cross the placenta. However, at the time of birth, when the placenta is pulled away, the baby's erythroblasts get into the mother's circulation. Once the immune system of the Rh- person "sees" the Rh antigen, antibodies to the Rh antigen will be made. During a second (or third, etc.) pregnancy with another Rh+ fetus, the antibodies can cross the placenta and destroy the fetal erythroblasts (baby blood cells), causing edema and a condition known as erythroblastosis fetalis or hemolytic disease of the newborn (HDN) and death or damage to the fetus. HDN can also occur when a mother is type O and the fetus is type A or B or when there is an incompatibility involving the "private" blood types discussed briefly below.
Now days, Rhogam (Rh gamma globulin) is administered to Rh- mothers immediately after the birth of an Rh+ baby. or after a miscarriage or amniocentesis. Usually the antibody titer of a pregnancy Rh- woman is monitored during the pregnancy. The Rhogam contains Rh antibodies gleaned from pooled blood of sensitized individuals (usually multiparous Rh- women). These antibodies attach to the fetal erythroblasts and get them "out of sight" of the mother's immune system. Rhogam is also to an Rh- woman given after a miscarriage or an amniocentesis to prevent sensitization.
Private Blood Groups
There are other blood groups which are considered "private" since there is one allele which is homozygous in most everyone and rarely does someone have a mutant allele (private allele). The P blood group is one of these and a patient who had this incompatibility was seen in our clinic. The woman, who had had several miscarriages, was pp (therefore, P antigen negative) and her fetuses were Pp (therefore, P antigen positive). The fetuses were all positive because her husband, like the majority of the population, was PP. Therefore, each pregnancy resulted in a Pp fetus. She then formed antibodies to the P antigens from her fetus's rbcs that escaped into her circulation. With each subsequent pregnancy the antibodies to the P antigen crossed the placenta and destroyed the erythroblasts of the fetus. Only heroic measures can help such pregnancies result in healthy live born babies.
Hemoglobinopathies
1. Hemoglobin which we will abbreviate as Hb, is the protein present in red blood cells responsible for oxygen transport. It is a tetramer made up of two dissimilar pairs of polypeptide chains (globin chains) and the iron containing prosthetic molecule, heme. The various subunits were cloned and sequenced between 1980 and 2000. There are a variety of mutations and, therefore, a variety of hemoglobinopathies. Hemoglobin electrophoresis can detect variants in individuals and DNA analysis can detect the more common mutations for carrier detection and prenatal diagnosis.
Protein Structure: Tetramer, four polypeptide chains. Two from alpha family and two from beta family of genes
2. Human Hb is heterogeneous. During development it is comprised of a succession of different globin chains which are differentially expressed during embryonic, fetal and adult life, i.e.,embryonic, fetal and 2 different adult forms.
Stage in
|
Hemoglobin |
Structure |
Proportion in
|
|
Embryonic |
Gower I |
zeta 2 epsilon 2 |
0 |
|
Fetal |
F |
alpha 2 gamma A 2 |
< 1% |
|
Adult |
A1 |
alpha 2 beta 2 |
97 - 98% |
Human Hemoglobins: Developmental appearances from embryo to adult
3. The disorders of Hb, the hemoglobinopathies, can be divided into two main groups, the structural variants (e.g. sickle cell HbS or HbC) and disorders of synthesis, the thalassemias. The former can be subdivided by the way in which they interfere with the normal function of Hb and/or the red blood cell, (e.g., abnormal oxygen affinity or hemolytic anemia). The latter can be subdivided according to which globin chain is synthesized abnormally, i.e., alpha, beta or delta-beta thalassemia.
Worldwide, 142 million people are carriers (heterozygotes) for Hb diseases and 250,000 severely affected (homozygotes) are born annually: 50% of these have Sickle Cell Disease (HbSS) and 50% have thalassemias.
4. Family studies of the various disorders of human Hb and analysis of the mutations responsible for these at the protein and DNA level have led to an understanding of the normal structure, function and synthesis of Hb. This has allowed demonstration of the molecular pathology of these disorders and prenatal diagnosis of a number of the inherited disorders of human Hb is possible.
5. Heterozygotes for hemoglobin mutations have about 60% protection from malaria. This is referred to as heterozygote selection.
6. The disorders of human Hb serve as excellent models for our understanding of the molecular basis of human inherited disease.
Brief overview of the various globin chains
1. The alpha globin chain family consists of the genes: zeta, alpha1 and alpha 2 (and pseudogenes)
2. The beta gene family consists of the genes: epsilon, gamma A , gamma G, delta, beta (plus pseudogenes)
3. Chromosome Locations: alpha gene family is on chromosome 16p and the beta family is on chromosome 11p
4. Gene Structure. Both families have 3 exons and 2 introns (common gene origin)
Evolution of the Hemoglobins:
Genes related to those found in hemoglobin have been found far back in the evolutionary tree. Over time, duplications and mutations of these related globin genes have occurred. The globin genes are, therefore, part of a much larger gene family. Because of the homologies, there have been misalignments among the alpha genes and also among the beta genes which are often the source of mutations. Misalignment within the alpha genes can cause deletion of an entire alpha gene which result in alpha thalassemias. Misalignments within the beta globin genes also lead to mutations such as the Lepore mutations which combine portions of the beta and delta genes and can cause beta thalassemia.
Unequal crossing over between the beta and delta globin genes cause mutations
There are two major groups of hemoglobinopathies
I. Structural Variants
HbS and HbC are two well known structural variants in beta globin genes. HbS (sickle cell hemoglobin) provided the first evidence for a single amino acid causing a genetic defect. The polypeptide sequence has a change from glu- to val0. A substitution mutation occurred in codon 6 and GAG (glu-) was changed to GTG (valo). HbC is another base substitution mutation in Hb beta gene codon 6 GAG (glu-) to AAG (lys+). Its effect on the hemoglobin molecule is less harmful. Sickle cell hemoglobin also provided the first evidence for non-overlapping code since only one amino acid was affected by the single base substitution mutation. The Hb beta gene was one of the first genes to provide evidence for introns when the mRNA was hybridized back to the DNA of the gene. Sickle cell disease is an example of how a single gene causes a multitude of problems. This is called a pleiotropic effect. Of interest is the fact that the difference between HbS and HbC phenotypes is significant.
Detection of Mutations. At first the HbS linkage to an RFLP site outside of gene (Founder Effect) was used to detect the mutation. Then the fact that the HbS mutation is a site for Mst enzyme was used. Now HbS and HbC are detected by PCR and Allele Specific Oligonucleotides (ASO) testing.
Newborn Screening. for hemoglobinopathies was begun in 1990 after the efficacy of penicillin prophylaxis was established.
Treatments used include: penicillin (to curb infections); hydroxyurea (to unmethylate fetal genes; bone marrow transplant (BMT) to have other gene source of beta globin.
II. Disorders of Synthesis: The Thalassemias
The thalassemias collectively are the most common human single gene disorders. They are due to decreased or absent synthesis of either the alpha or beta globin. The pathophysiology is due to a distortion of the alpha/beta chain ratio: the non-mutant chain, produced in relative excess, forms a tetramer (e.g., alpha 4 or beta4). The excess precipitates in the cell, damages the membrane and leads to premature destruction of the rbcs. In addition, the defect produces a hypochromic, microcytic anemia due to a reduced amount of Hb. The prevalence of Hb mutations is presumably is due to the protective advantage that heterozygotes have against malaria (heterozygote advantage). Compound heterozygotes are common since many mutations can reduce the synthesis of either the alpha or beta chain, and, in addition, there are many structural mutations that may be combined with a thalassemia mutation. Many complex hematological problems result from the many possible combinations of mutations.
1. ALPHA THALASSEMIA
Alpha thalassemia is the reduction or absence of alpha chain synthesis. Look in your textbook for a figure of the alpha genes on chromosome 16. There are two functional alpha genes: alpha 2 produces 2 to 3 times more chains than its downstream partner, alpha 1.
Mutations. The most common cause of alpha thalassemia is a deletion of one or more alpha globin genes. This is one of the few times when a gene defect is due to the total deletion of a single gene. Other types of mutations do exist, however. Deletions are caused by misalignment due to extended homology of the two duplicated alpha genes (alpha1 and alpha 2) and surrounding DNA. This is verified by the occasional finding of a person with three copies of the alpha genes on one chromosome. Although the alpha thalassemias are found worldwide, the homozygous deletion of all four alleles is largely restricted to Southeast Asians. This is because they have a higher frequency of the - - haplotype in carriers. This is referred to as a Founder Effect. The - - haplotype frequency is actually greater than the normal alpha alpha haplotype in some Melanesians and certain East Indians.
Clinical features. Because alpha globin is found in both fetal and adult hemoglobins, this disease causes intrauterine as well as postnatal disease. Infants with severe alpha thalassemia and high levels of Hb Barts (gamma 4) suffer severe intrauterine hypoxia and are born with massive generalized fluid accumulation, hydrops fetalis. In milder alpha thalassemias, an anemia develops because of the gradual precipitate of the HbH (beta 4) in the erythrocytes leading to their premature destruction.
Table of Clinical States of Alpha Thalassemia Genotypes (note clear gene dosage effect)
| Clinical Condition | Number of alpha genes |
Genotype | alpha chain production |
Clinical disorder |
| Normal | 4 | ++/++ | 100% | none |
| Silent carrier | 3 | ++/+ - | <75% | none |
| alpha thalassemia trait | 2 | - - / ++ SE Asia | 50% | mild anemia, microcytosis |
| HbH (beta4) disease | 1 | + - / - - | 25% | moderately severe hemolytic anemia/ variable anemia and splenomegaly |
| Hydrops fetalis Hb Barts (gamma4) | 0 | - - / - - | 0% | death in utero, profound anemia |
Diagnosis: --/-- homozygotes for alpha thalassemia 1 show 80% Hb Barts (gamma 4) and a persistence of embryonic hemoglobins (zeta and epsilon); + - /- - patients with HbH (beta 4) disease show reduced MCHC (mean cell hemoglobin concentration) and reduced MCV (mean cell volume) with 5-30% HbH (beta 4) and some Hb Barts (gamma 4). Alpha globin synthesis is reduced or absent.
2. BETA THALASSEMIA
Beta thalassemia is a reduction or absence of beta globin chain synthesis; it is also known as Cooley's Anemia, beta thalassemia major, Mediterranean anemia, homozygous beta thalassemia major. Unlike alpha globin, beta globin chains are important only in the postnatal period and, therefore, their loss is not apparent until a few months after birth when the beta globin normally replaces the gamma globin. There is also thalassemia intermedia which is a milder form where the individual makes some beta chains. Beta thalassemia minor refers to the carrier or heterozygote. Carrier frequency is 1/10 Italians and Greeks; 1/25 Asians (Chinese); 1/50 American Blacks. Heterozygotes have smaller rbcs and a smaller than normal MCV < 70.
Mutations. In contrast to alpha thalassemias, the beta thalassemias are usually due to base pair substitutions rather than deletions. There is such a variety (more than 100 are known) of these mutations that most persons carrying two mutations are compound heterozygotes. The symbol 0 denotes beta thalassemias in which so little beta globin is produced that no HbA is present. The symbol + denotes beta thalassemias in which some beta chain is made and Hb A is detectable.
The Molecular Basis of Beta Thalassemias
Mutations that affect the levels of mRNA beta+ mutants
- promoter mutants and RNA splicing mutants, the most common and most interesting. These were the first such mutations documented.
- splice junction mutation in the 5' or 3' acceptor sites or consensus sequences (GT....AG) cryptic splice sites in either an intron or exon
- HbE dual effect--nucleotide/amino acid substitution which activates a cryptic splice site and moderately reduces the amount of mRNA
- Also an example of a silent mutation that is not neutral in its effect is codon 24 GGT to GGA both code for glycine but the mutation activates a cryptic donor site because it can be misread.
- mRNA capping and poly A mutants
- Other mutations that do not affect levels of mRNA can affect protein stability. These defects may be in the coding regions: nonsense and frameshift mutations lead to short, unstable polypeptide chain.
- Gene deletions are an infrequent cause. The only common deletion is a 3' 619 bp deletion in Asian Indians. Hb Lepore is a structural variant due to unequal crossing over between the beta and delta genes resulting in a fusion protein that is unstable.
- Beta° thalassemia mutations are due to deletions and nonsense or frameshift mutations which completely knock out the synthesis of beta globin. Beta+ thalassemia mutations are due to the wide variety of donor or acceptor splice site or cryptic site mutations in introns and exons which lead to varying decreases in the output of beta globin chains. They can also be caused by mutations in several upstream promoter regions. The large number of different mutations results in people who are compound heterozygotes exhibiting a wide variety of phenotypes. Some mutations, because of a founder effect and/or heterozygote selection, result in specific ethnic distributions of alleles. Since some mutations are quite frequent in certain populations, carrier detection is possible to determine the risk of disease.
Heterozygote or carrier detection is done on a large scale in the Mediterranean. The screening program was similar to that for Tay Sachs Disease in the Jewish community. In 1977 a pilot program for carriers in Southern Sardinia was begun using an accurate and inexpensive blood test showing reduced rbc volume (MCV < 70) and relatively increased HbA2 compared with HbA due to reduction in amount of alpha 2 beta2 relative to alpha 2delta 2. Public education was a large component. Ninety-five percent of Sardinians with beta thalassemia have the mutation in codon 39 which is a beta 0 mutation (no beta chain made, a null allele). The CAG (glu) to UAG (stop) results in a 39 amino acid beta chain instead of the normal 146 amino acid chain. Two percent is due to a frameshift at codon 6 which gives a truncated protein and some are beta+ splicing mutations: nucleotide 110 in intron 1 and 745 in intron 2. These three mutations account for 98% of the beta thalassemia mutations in the Sardinian population.
Clinical phenotypes show much variability among homozygotes or compound heterozygotes. Some individuals have profound anemia and are total transfusion dependent and their life span is reduced despite supportive measures. Other individuals have moderate anemia and occasional transfusion is required and others have only mild anemia where no transfusion is required. In beta thalassemia, HbF is often increased and HbA2 may be reduced, normal or increased. Elevation of HbA2 is unique to beta thalassemia heterozygotes. HbF is increased because of selective survival of cells making HbF. Heterozygotes with only one beta globin gene mutation may have a mild anemia,"silent" changes with no hematological change, or severe anemia ("dominant" inheritance) if they have a single null mutation.
Genotype-Phenotype Correlations. Mild phenotypes are associated with mutations that lead to truncated chains up to ~ 72 residues. Presumably these short fragments are degraded. Severe phenotypes are associated with mutations in exon 3 which result in longer truncated products, or elongated and unstable chains. These can bind heme and have secondary structure which makes them less susceptible to proteolytic degradation. HbS/beta thalassemia combination is worse than the homozygous state of either allele. The symptoms are more like sickle cell disease in these compound heterozygotes.
HbE is common in the southeast Asian islands. The mutation opens a cryptic splice site and results in the reduction of synthesis of the beta chain. A compound heterozygote for HbE and a beta thal mutation leads to variable phenotypes from normal to the equivalent of a severe beta thal homozygote phenotype.
Many modifier genes have been found which contribute to the variable expression of thalassemias. As is true for many genetic disorder, it is not just simple mendelian genetics.
Hereditary Persistence of Fetal Hemoglobin.
The basis for the variable persistence of Hb F production in beta thalassemia is not fully understood. In some cases, there are point mutations in the promoter regions of the globin genes or major deletions involving the beta globin gene cluster. In others, there is no obvious reason for the unusually high level of gamma-globin production in response to anemia (possibly involving other regulatory loci?). The deletions that include the 5' end of the beta globin genes or point mutations in their promoter regions are associated with higher levels of HbF. These are of particular interest because they impair the perinatal switch from gamma to beta globin synthesis.
Symptoms and Treatment
Untreated thalassemia results in heart failure from severe anemia, severe enlargement of the liver and spleen, retarded growth, and changes in the bones. Symptoms involve many organs and usually there are problems in puberty. The less severe thalassemia intermedia has a later onset.....later than 2 years of age....it may manifest by 7 years or not until adulthood.
Transfusions are a common treatment. However, complications from transfusions include hepatitis, AIDS, antibody formation, and iron overload--10-20 mg/day. Iron overload poisons the heart, liver, and other organs. To circumvent this iron chelators or splenectomy may be used. BMT (bone marrow transplant) is another (risky) option.
Summary
The thalassemias, the most common monogenic diseases in humans, and the first to be analyzed at the molecular level, show remarkable phenotypic heterogeneity. Much of the variability can be ascribed to the pathology due to complications of the profound anemia. And recently some progress has been made in the molecular basis for phenotypic variability of the thalassemias. In this brief over view of the hemoglobinopathies we noted that there is a vast array of mutations as well as both locus heterogeneity(all the different a and b globin family genes) and allelic heterogeneity (all the myriad of mutations in each globin gene).
A Sickle Cell and Thalassemia Lecture
Osteogenesis Imperfecta: A Skeletal Disorder
There are many types of collagen (Type I - XIV) in the body and each have their own COL genes. Collagens form part of the extracellular matrix of connective tissue including bone and cartilage. The COL genes also belong to a gene family whose members are dispersed around the genome (unlike the beta globin gene family which are close together on chromosome 11p and the alpha globin genes on chromosome 16p). Like the globin gene mutations, however, there are quantitative mutations which cause less of one of the polypeptides to be made and qualitative mutations which change the structure of the polypeptide. The translation, assembly and subsequent post-translational modifications of the subunits of collagen are complex. Mutations can affect any step in the process.
The mutations that result in OI are in the genes COL1A1 on chromosome 17 and COL1A2 on chromosome 7 which code for the two polypeptides (alpha 1 and alpha 2) of which collagen type I is composed. Collagen type I is composed of two alpha 1 and one alpha 2 polypeptide chains. There are several types of OI..... type I, II, III and IV.....all of which are autosomal dominant. There was an idea at one time that there were autosomal recessive forms because sibs with normal parents were affected. We now know that they are new spontaneous mutations with gonadal mosaicism. The sperm of one unaffected father with two affected offspring was analyzed and a high proportion of his sperm was shown by DNA analysis to carry a mutation in one of the COL genes. (N.B. Do not confuse the clinical types I, II, III, IV with the Collagen types I, II, III, etc.)
Pedigree illustrating the inheritance of Osteogenesis imperfecta due to a new mutation and germ line mosaicism in the father
1. OI type I is due to a null allele (one which has no protein product) of COL1A1 and it results in a less severe clinical picture since the collagen that is made is normal there is just less of it. The phenotype is one of blue sclera, brittle bones but no bony deformity, normal stature and often pre senile deafness.
2. OI Type II is a perinatal lethal form which results in very early death. Abnormal collagen is made because of missense mutations in COL1A1 or COL1A2 which substitute other amino acids for gly toward the C terminus. (Glycine is one of the amino acids which are modified during post translational modification). The fetus has severe skeletal fractures and deformities and dark sclera. Since this is a genetic lethal new cases arise exclusively from new mutations.
3. OI Type III is a progressive deforming form also due to abnormal collagen caused by missense mutations in COL1A1 or COL1A2 which substitute another amino acid for gly, mostly towards the N terminus. At birth there are often fractures and bony deformities, there are blue sclera, dentinogenesis imperfecta, hearing loss, and severe scoliosis (dwarfed).
4. OI Type IV is another milder form. It is caused by missense mutations in glycine codons for the COL1A1 and COL1A2 genes and exon skipping mutations in the 5' end of the COL1A2 gene. The phenotype is of normal colored sclera, mild to moderate bony deformity, short stature, fractures, (sometimes confused with child abuse) hearing loss, dentinogenesis imperfecta and obvious bruising. (This is probably what Toulouse Latrec, the French cabaret artist suffered from.)
In the prenatal diagnostic center, we saw a young couple (he was 16 years old and she was 15 years old) who were referred to us due to an abnormal ultrasound at her clinic. At first we thought the fetus had achondroplasia and she was referred to Cedars-Sinai to the skeletal dysplasia team for a second opinion. The fetus was seen to have a severe skeletal dysplasia and all of the bones except one humerus appeared to be bent or broken. The skull was abnormal and ribs were abnormal in shape and the skull showed abnormal density. The opinion was that the fetus had OI. A second ultrasound at Cedars showed obvious breaks in all long bones, the fetus was extremely small, and it had a compressible skull. The diagnosis was Lethal Osteogenesis Imperfecta (OI Type II). The baby was not expected to live through the birthing process but he did. After 28 days in the hospital the baby was taken home by the parents. The diagnosis was changed to OI Type III (because it lived). After a serious but futile attempt on the part of the young parents to care for the baby, the baby was placed in an adoptive home so that it could receive the type of care it required. Since neither parent was affected, this case is the result of a new spontaneous mutation in one of the two teenage parents.
Receptor and other Membrane Protein Defects
FGFR Mutations
There are several human skeletal disorders caused by fibroblast-growth-factor receptor (FGFR) mutations. There are different genes that code for various FGFRs and mutations which result in clinical syndromes occur in all of them. The FGFRs all belong to a family of tyrosine kinase cell membrane receptors and although they represent a gene family, each is coded for by genes at different loci and on different chromosomes. These FGFRs have Ig-related extracellular domains, a membrane spanning domain, and an intracellular tyrosine kinase domain. Mutations can affect any of these domains. The individual polypeptide gene products dimerize when they bind their specific fibroblast growth factor and if there is a mutation in one allele, both normal and abnormal receptors are made which cannot associate and dimerize as well. Mutations can also affect ligand (growth factor) binding. Mutations have been mapped to FGFRs 1, 2 and 3 for the several clinical syndromes they cause. Mutations in the same FGFR gene or in different FGFR genes can give rise to the same clinical phenotype (e.g., clinically defined Pfeiffer syndrome can be due to mutations in either FGFR1 or FGFR2). Different mutations in the same gene can also give rise to the same clinical phenotype (e.g., thanatophoric dysplasia can be caused by different mutations within the FGFR3 gene). Hypochondroplasia, achondroplasia, thanatophoric dwarfism, Pfeiffer syndrome, Apert syndrome Jackson-Weiss syndrome, and Crouzon syndrome are all caused by mutations in FGFR genes.
During a prenatal counseling session for advanced maternal age, the woman was noted to have a somewhat normal variation of head shape. When her baby was born it had craniosynostosis and surgery was considered to prevent brain damage (it was later deemed unnecessary). The birth of the more severely affected baby prompted a better look at the entire family. The mother and six out of her eight children had prominent coronal protuberances and/or irregular or crowded teeth but their hands and feet were normal (no synostosis). It is highly probable that this condition is a variant of Crouzon syndrome and is due to a mutation in an FGFR gene.
Crouzon Disorder
Another case in the pediatric unit of the hospital was a 22 month old patient diagnosed with Crouzon disorder who had cranioplasty because of her craniosynostosis. The child might have Jackson-Weiss which has all the same features as Crouzon but has foot anomalies. This child had a very large big toe on one foot. Crouzon is usually distinguished from other similar FGFR disorders involving craniosynostosis (Pfeiffer, Jackson-Weiss, Apert) by lack of hand or foot anomalies. The family history was unremarkable, therefore the child represented a new mutation in one of her parents (most likely the father). The recurrence risk would be 0% if the mutation occurred in one gamete. But if the mutation occurred in a "feeder" cell in the gonad it would give rise to many gametes with the same mutation. This result is called gonadal mosaicism (gonad contains both normal and mutant gametes) and the recurrence risk is greater than 0%. Although it is not possible to know for certain what the recurrence risk is, a risk of 0 to 7% has been given in some cases.

Pfeiffer Syndrome
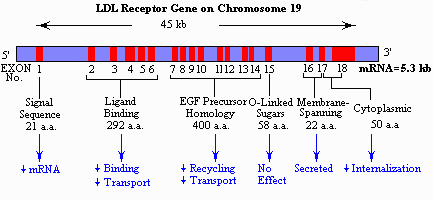
Hypercholesterolemia (FH)
Familial hypercholesterolemia (FH) is caused from a defect in the gene for low density lipoprotein (LDL) receptors. LDL is a protein-lipid complex. Normally cholesterol enters the cell by endocytosis via clathrin coated receptors. The resulting endosomes with the LDL are merged with lysosomes and the protein and the lipid portion of LDL are hydrolyzed by lysosomal enzymes to their subunits, amino acids, cholesterol and fatty acids. Normally, this exogenous (brought in from outside the cell) cholesterol inhibits the activity of cellular enzymes involved in endogenous (synthesized within the cell) cholesterol synthesis so that there is a steady-state level of cholesterol in the liver.
When the system fails, there is too much cholesterol circulating and the individual develops atherosclerosis and eventually has a heart attack. This condition is inherited in an AD pattern since half the normal number of LDL receptors is insufficient to prevent heart attacks by 35 - 40 years of age and death (particularly of males) by 60 years. There is a clear gene dosage effect since homozygotes may have clinically significant coronary heart disease in childhood and few live beyond the third decade. The clinical signs include collections of fat in xanthomas at the elbows, knuckles and around the periphery of the cornea and atherosclerotic plaques. FH is not responsible for the majority of heart attacks in the general population. Homozygotes occur 1 in a million but the heterozygote incidence of the FH is 1/500. The treatment for FH is bile acid depletion by a binding resin (cholestyramine and colestipol) and HMG CoA reductase inhibitor (lovastatin).
There are several classes of mutants in FH. The classes of mutations are correlated with the region of the gene mutated. The receptor protein has five domains, four of which correspond to the 4 classes of mutants that have been found. Alu repeats found in several introns as well as an Alu-like sequence in exon 13 are the cause of deletions.
Class 1 mutations are those that result in the deficiency of the receptor due to lack of transcription or translation.
Class 2 mutations result in the improper folding of the protein with the consequent trapping of the receptor in the endoplasmic reticulum (ER).
Class 3 mutations result in improper binding of LDL to the LDL receptor protein.
Class 4 mutations result in internalization defects due to improper clustering of the receptors in the coated pits.
Cystic Fibrosis
Cystic fibrosis (CF) is an AR disorder due to a mutation in the cystic fibrosis trans membrane protein (CFTR) which regulates chloride (Cl-) transport into the cell. The gene for CFTR is on 7q31.3. Carrier frequency among Caucasians of European descent is about 1/22; 1/46 among Hispanic Americans; 1/66 among African Americans; and 1/150 among Asian Americans.
The disease is progressive and affects the bronchial system and the gastrointestinal tract. It also affects the vas deferens in males.
The most common mutation in all ethnic groups is a 3 base pair deletion, F508, which deletes phenylalanine at the 508th amino acid/codon in the normal protein. Over 800 mutations have been identified at this locus. The different mutations have different frequencies in different populations. Screening for carriers is possible using allele specific oligonucleotides (ASOs) for each of the common mutations. However, carrier screening is controversial because although you can detect the majority of mutations (85%), you cannot be certain a person is not heterozygous for some less common mutation that is not in the mutant screening panel. Some laboratories are introducing sequencing to detect rare mutations.
For DNA diagnosis of the known mutations, regions of the CFTR gene are amplified by PCR, then hybridized to specific CF mutation oligonucleotide pools (ASOs). A child suspected of having CF because of clinical symptoms can be tested for the kind of mutations s/he has. The cells used in the test are obtained by a small brush that picks up cheek cells from the inside of the individual's mouth. Some CF fetuses have been detected by prenatal ultrasound due to an echogenic bowel. The echogenic bowel is believed to be due to a meconium plug often found in CF babies. Once the mutations in an affected child are known, the parents can have DNA analysis and future pregnancies can be monitored by chorionic villus sampling or amniocentesis and ASO analysis. Congenital bilateral absence of the vas deferens (CBAVD) occurs in 95% of male patients with CF and may be the only clinical symptom in some males with specific mutant alleles. Idiopathic chronic pancreatitis and increased respiratory problems are also associated with mutations in the CFTR gene.

Classification Scheme for CFTR Mutations Mutation Class Effect of Mutation on CFTR Protein Mechanisms
| Mutation Class |
Effect of Mutation on CFTR Protein
|
Mechanisms
|
|
I
|
Reduced or absent synthesis
|
Nonsense, frameshift, or splice-junction mutations
|
|
II
|
Block in protein processing
|
Missense mutations, amino acid deletions
|
|
III
|
Block in regulation of CFTR chloride channel
|
Missense mutations
|
|
IV
|
Altered conductance of CFTR chloride channel
|
Missense mutations
|
Ten Most Common CFTR Mutations in Caucasians and their Phenotypic Expression
|
Mutation
|
Relative
Frequency |
Mutation
Functional Class |
*Pancreatic
Sufficient (PS)/ Insufficient (PI) |
|
delta F508
|
66.0%
|
II
|
PI
|
|
G542X
|
2.4%
|
I
|
PI
|
|
G551D
|
1.6%
|
III
|
PI
|
|
N1303K
|
1.3%
|
II
|
PI
|
|
W1282X
|
1.2%
|
I
|
PI
|
|
R553X
|
0.7%
|
I
|
PI
|
|
621+1G->T
|
0.7%
|
I
|
PI
|
|
1717-1G->A
|
0.6%
|
I
|
PI
|
|
R117H
|
0.3%
|
IV
|
PS
|
|
R1162X
|
0.3%
|
Not clear probably II: Transcript is stable; truncated protein is probably mis folded |
PI
|
* Degree of pancreatic function in patients with cystic fibrosis. PS mutations are dominant to PI mutations; thus, patients with PS/PS and PS/PI genotypes are usually pancreatic-sufficient, whereas patients with PI/PI genotypes are usually pancreatic-insufficient.
Duchenne and Becker Muscular Dystrophies
Duchenne and Becker muscular dystrophies (DMD and BMD) are XR disorders due to mutations in the gene for dystrophin. Dystrophin is a protein found in skeletal and cardiac muscle. Dystrophin has several distinct domains including an actin binding domain and a spectrin-like repeat domain. Spectrin, a long fibrous protein, is the primary component of the erythrocyte cytoskeleton. Becker alleles produce a much milder phenotype than DMD. In DMD there is little or no dystrophin, whereas in most BMD patients the protein is present at reduced levels. The gene is very large, 2300 kb, and as a consequence, like the NF1 gene, it has a a high mutation rate. Although point mutations can give rise to DMD or BMD, 60% of the mutations are large duplications or deletions. These duplications and deletions are due to misalignment of the homologs during recombination in meiosis. The misalignment is due to the fact that the gene contains repeating sequences that code for the repetitive protein domains. BMD mutations have been found to be "in frame" deletions (or duplications) where the reading frame is preserved and an internally deleted but partially functional protein product results. DMD mutations, on the other hand, have been found to be due to deletions (or duplications) that result in an "out of frame" or frameshift mutation and the inevitable encountering of a stop codon after a short distance. The resulting protein will be truncated and degraded in the cell (also similar to the NF1 gene product).
Females are usually not affected with DMD because DMD is a genetic lethal and since affected males do not reproduce. One-third of cases are predicted to be due to new mutations. Occasionally, a female carrier of the gene is affected because of a skewed X inactivation pattern which results in a large percent of her muscle cell nuclei having the normal allele inactivated. This situation is rare although 8% of carriers exhibit some muscle weakness. Successful myoblast implantation from healthy mice to mutant mice with mutations in dystrophin showed some promise. However, in humans the transplants have been less effective.
A protein called utrophin, an homologous protein, is able to compensate for the absence of dystrophin and prevent the development of symptoms in a mouse model of DMD. The utrophin gene contains two promoters, one of which might make a good target for drugs designed to increase its activity. It may be possible in the future to increase the expression of utrophin in the cells of DMD patients to the point where it could counteract the symptoms of the disease.
Enzyme Deficiencies
Genetic disorders involving enzyme deficiencies are usually inherited in an AR pattern. Because of the efficiency of enzyme activity, the 50% of an enzyme activity provided by the one functional allele in a heterozygote is usually sufficient. This is an example of gene dosage. One allele makes 50% of the amount of two alleles.
There are other obvious examples of gene dosage effects as found in the globin alleles, polygenic-additive traits such as height, and also some cancer cells over express by gene amplification (i.e., double minutes which are very small accessory chromosomes and homogeneously staining regions, HSRs) with a concomitant increase in the cell product.
The problems caused by enzyme deficiencies can be due to the buildup of substrate(s) and/or the lack of the product(s) in the metabolic pathway in which the enzyme is participating. The inborn errors of metabolism, if left undetected and untreated, end in serious consequences (brain and other organ damage) and often death in the infant.
E1 E2 E3 E4
A------------B-----------C-----------D------------E
S P/S P/S P/S P
E = enzyme S = substrate P = product
Mutations in enzymes have helped biochemists piece together the steps of metabolic pathways. Beadle and Tatum won the Nobel Prize for their work on mutants of the fungus, Neurospora (a bread mold). They coined the expression, "one gene, one enzyme."
Acute Intermittent Porphyria (AIP)
Acute Intermittent Porphyria (AIP) is an exception to the rule that enzyme deficiencies are AR or XR, since AIP shows an AD pattern of inheritance. The enzyme that is deficient in (AIP) is porphobilinogen deaminase (PBG deaminase). This is an important enzyme in the heme synthesis pathway. Normally, the person carrying the mutant gene is healthy because 50% of enzymatic activity is enough to keep the pathway going. However, precipitating factors in the environment can cause problems by lowering the concentration of heme in liver cells. Drugs such as barbiturates, some steroid hormones and alcohol can cause an increase in synthesis of the the P450 hepatic cytochromes which are detoxifying enzymes in the smooth endoplasmic reticulum (SER). Cytochromes contain heme and when they are synthesized in response to environmental triggers, the level of heme is lowered at that time. Heme is normally involved in a feedback inhibition of delta-aminolevulinic acid synthase (ALA synthase) and when the heme concentration is reduced, there is an increase in the level of the transcription and translation levels of ALA synthase. The increased amount of the ALA synthase then causes an increase in the level of its products, ALA and PBG. Under these circumstances, the AIP heterozygote, with only ½ the normal level of PBG deaminase, cannot handle the increased amounts of ALA and PBG. These high levels of ALA and PBG cause the clinical symptoms of the disease. The symptoms are as diverse as abdominal pain and psychosis. The peripheral and central nervous systems are both affected. King George III, the "mad" king, is believed to have suffered from a related disorder, porphyria variegata. This disorder is also autosomal dominant with reduced penetrance. A different enzyme in the porphyrin pathway, protoporphyrinogen oxidase (PPOX), the penultimate enzyme in the heme biosynthetic pathway, is responsible. He is said to have had the clinical features consistent with the diagnosis: rapid pulse, fever, yellow or bloodshot eyes, abdominal colics, constipation, cramps in the legs, skin blistering on his arms, port-wine-colored urine, and rambling speech degenerating into obscenities and hallucinations. Some of his ancestors are known to have suffered with similar problems.
ALA synthetase * PBG deaminase
glycine + succinyl CoA ------------------------ ALA---------------------------------- PBG-----------------hydroxymethlbilane---heme
Phenylketonuria, tyrosinemia, albinism, alkaptonuria
Urea Cycle Disorders
CPSI (Carbamoylphosphate synthetase I deficiency)
OTC (ornithine transcarbamylase deficiency)
ARG argininemia (arginase deficiency)
ASL Argininosuccinic aciduria (argininosuccinic acid lysase deficiency)
ASS Citrullineamia (argininosuccinic acid synthetase deficiency)
NAGS (N-acetyl glutamate synthetase deficiency)
The lysosomal storage diseases
The lysosomal storage diseases are a group of disorders (inborn errors of metabolism) due to deficiencies in the one of the multitude of acid hydrolases found in the lysosome. Lysosomes, as you have learned in cell biology, are ubiquitous, single membrane bound organelles that originate from the golgi body. The lysosomal enzymes are selectively packaged into lysosomes using a special zip code" of mannose-6-P residues added by enzymes in the golgi body. These residues are recognized by receptors in the newly forming golgi derived lysosomal membranes. The role of lysosomal enzymes is to breakdown "old" molecules and organelles and to do this they contain a wide variety acid hydrolases able to break down all macromolecules including polysaccharides, proteins, lipids, nucleic acids. They break their substrate down one subunit at a time. Several enzymes are usually involved in the sequential breakdown of any one substrate such as oligo- and polysaccharides. The symptoms of the various lysosomal storage diseases depend what enzyme is missing, its function, and its site of action (where is the missing enzyme's substrate? --joints, liver, brain, etc.).
Mucopolysaccharidoses (MPS)
Mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases which are due to the loss of enzymes that break down the polysaccharides of the proteoglycans (also known as, mucopolysaccharides) found in the extracellular matrix. Hurler and Scheie are due to mutations in the same gene, alpha-L-uronidase, and belong to the same complementation group. However, the Hurler mutation(s) cause a more devastating clinical picture. The compound heterozygote (H/S) is clinically less affected than the homozygous Hurler and more affected than the homozygous Scheie. Carrier detection is based on being able to measure the enzyme activity of homozygous normal, heterozygotes and homozygous mutants and distinguish among them. The enzyme activity of the three genotypes fall into three fairly distinct groups but not without some overlap. Hunter is another MPS which is X-linked. The clinical picture is similar to Hurler but a different lysosomal enzyme is involved.
Tay Sachs Disease (TSD)
Tay Sachs Disease (TSD) is an AR lysosomal storage disease due to a deficiency or absence of the lysosomal enzyme, hexoseaminidase A. It causes severe mental and physical deterioration usually beginning in infancy. Death usually occurs between 2 and 3 years of age. TSD is another example of an ethnic disease with a higher incidence among Ashkenazi Jews and French Canadians (1/30 as opposed to 1/170 in non Ashkenazi Caucasians). The disease affects primarily the CNS and the brain specifically. The infantile form manifests itself with progressive loss of function associated with the death of brain cells engorged with the substrate of the missing lysosomal enzyme. The missing enzyme is hexoseaminidase A which is responsible for the cleaving of a N-acetyl galactose (a hexose) from its substrate the glycosphingolipid GM2 ganglioside, so named because of its mobility on a gel. The sphingolipid substrate is found primarily in the brain and CNS where it forms part of the myelin sheath. This explains the clinical course of the disorder.
TSD carrier testing in the Ashkenazi Jewish population has been a large success much like the thalassemia carrier testing in the Mediterranean populations (see earlier discussion). TSD has been reduced by 95% in the Ashkenazi Jewish population. During the last 25 years more than a million people have been tested. The test is an enzyme assay for hexoseaminidase A activity and it is performed on blood leukocytes for carrier detection and on chorionic villi cells or amniocytes for prenatal testing. Once the condition is detected, the specific mutation(s) can be determined by DNA analysis for prognosis and for future prenatal detection in that family. Because of the blood brain barrier, enzyme therapy for TSD has proven totally unsuccessful. Hyperbaric oxygen treatment has been attempted but it has not proven successful.
Due to allelic heterogeneity, there are infantile, juvenile and adult onset forms of the disease. Three mutations account for 98% of the mutations in the Ashkenazi Jewish population. The two which are responsible for the infantile form of TSD result in no enzyme activity and are, therefore, null alleles. One is a 4 base pair insertions which causes a frame shift mutation. The other one which causes infantile onset occurs at the 5' border of intron 12 and changes a G to a C. The 5' border of introns has a consensus splice donor sequence, GT. The 3' borders of introns also have a consensus splice acceptor sequence, Since the spliceosome complex must recognize the consensus sequences, a mutation in the 5' or 3' sequence leads to failure of splicing at the correct site. The splicing machinery must then scan farther until a "cryptic" donor (or acceptor) sequence is found. As a consequence some intron sequence is included in the mature mRNA and translation of this inserted sequence leads to an abnormal segment of the protein and usually a stop codon is encountered due to the frameshift and a truncated protein results. The common adult onset mutation is due to a base substitution in codon 269 resulting in a serine replacing a glycine residue in the enzyme. The result is a greatly reduced amount of the enzyme. Infantile onset occurs when the individual has two null alleles, juvenile onset occurs when the individual has one null and one adult onset allele, and adult onset occurs when the individual has two adult onset alleles.
A 32-year-old (1998) CSUDH student who was French Canadian had juvenile onset TSD. He had one null allele, the 4 base pair insertion (+TATC) from his mother and the other allele from his father is the Gly269Ser base substitution mutation. His hexoseaminidase A enzyme level is 4.7%. He had profound muscle weakness and was confined to a wheelchair. Four of his nine siblings had juvenile onset TSD.
TSD also shows locus heterogeneity because hexoseaminidase A is composed of an alpha subunit, a beta subunit, and it also requires an activator protein which binds the ganglioside substrate to present it to the enzyme. The gene for the alpha subunit is at 15q23-24. The gene for the beta subunit is on chromosome 5 and the gene for the activator protein is also on chromosome 5. Hexoseaminidase B is formed of two beta subunits and homozygosity for mutations in the beta subunit gene cause Sandhoff disease in which neither hexoseaminidase A or B are made.
I-Cell disease
I-Cell disease was so named because of the "inclusion" bodies (engorged lysosomes) found in the cells of the affected individuals. Although it is clearly a monogenic AR disorder, multiple lysosomal enzyme activities are lost. The phenotype is consistent with the loss of activity of many lysosomal enzymes. The disorder shows a range of pleiotropic effects including abnormal facies, skeletal changes, severe growth and mental retardation. Survival is only for 5 to 7 years. Like many gene mutations before this one, the study of this disorder led to the discovery of a cellular pathway, namely how lysosomal enzymes are packaged by the golgi for incorporation into lysosomes. The defect in this disorder is in the golgi body enzyme, phosphotransferase, (chromosome 4) responsible for the addition of the recognition signal, mannose-6-P, necessary for the uptake of all lysosomal enzymes into lysosomes.
Enzyme replacement therapy
Enzyme replacement therapy for lysosomal storage diseases disorders has not been particularly successful because of the difficulties in delivery of the enzyme to the target cells and/or target organelles. However, Gaucher Disease (AR), the most prevalent lysosomal storage disease, has been a model system for enzyme replacement therapy (ERT). Incidentally, Gaucher is even more prevalent in Ashkenasi Jews than TSD. Gaucher is due to a deficiency of glucocerebrosidase, a lysosomal enzyme which degrades a glucocerebroside another complex lipid. The gene is located on chromosome 1q21. There is allelic heterogeneity with some mutations causing more severe symptoms than other alleles and many who are affected are compound heterozygotes. The variability in the phenotype is remarkable ranging from severely affected infants to asymptomatic adults. Many patients suffer from anemia, bone damage, and swelling of the liver and spleen, while a few develop severe CNS damage and death. All have a defect in the same enzyme. The reticuloendothelial system is the most affected and the lysosomes of macrophages in the liver, spleen, and bone marrow become engorged with the substrate. The consequence is the reduction in the number of RBCs and platelets. Bone lesions and osteonecrosis cause pain and general illness. Some few patients have nervous system degeneration.
Traditionally, Gaucher disease was managed by supportive therapy including total or partial removal of the spleen, transfusions, orthopedic procedures and occasionally bone marrow transplantation (BMT) (see below). Enzyme replacement therapy has successfully reversed many of the manifestations of the disorder including correcting blood counts and reducing organ size in many patients. The enzyme can be purified from human placenta and chemically modified (alglucerase) to expose mannose residues needed for internalization by macrophages which have mannose receptors in their cell membrane. However, the therapy is quite costly, often ranging from $100,000 to $300,000 for one year for an adult patient. Researchers are still investigating the optimal doses of enzyme preparation.
Recently ERT has been implemented for Fabry, Hurler, and Pompe Diseases on a trial basis.
Bone marrow transplantation (BMT)
Bone marrow transplantation (BMT) has been tried for lysosomal storage diseases including Gaucher. The transplanted cells are a source of lysosomal enzymes that can be transferred to other cells through the extracellular fluid. Donor bone marrow stem cells give rise to cells (mononuclear-phagocyte system) that travel to many tissues throughout the recipient's body. The enzyme replacement therapy is highly effective in reducing bone pain, the size of the liver, spleen and heart and there have even been a few reports of positive effects on nervous system damage in some storage diseases.
Peroxisomal Disorders
Peroxisomes are single membrane bound organelles in the cytoplasm of most, if not all, eukaryotic cells. They contain an array of enzymes which can vary with the type of organism and the type of tissue they are in. However, they all contain essential enzymes that carry out important oxidation reactions. Examples of peroxisome reactions in humans are oxidation of fatty acids, degradation of phytanic acid (from plants) and synthesis of plasmalogens. (It is believed that peroxisomes arose by endosymbiosis of prokaryotes which could detoxify O2 for their early eukaryotic host cells.)
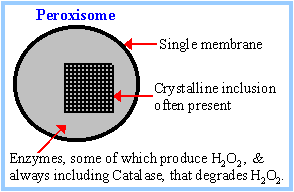
Peroxisomal proteins, unlike lysosomal proteins but like mitochondrial proteins, are synthesized on free cytoplasmic ribosomes and not in the ER. The way in which the peroxisomal proteins find their way to a peroxisome is via peroxisomal targeting signals (PTS) which are recognized by PTS receptors. There are two known PTS's, one type is at the C-terminus of a peroxisomal protein (PTS1) and one is at the N-terminus of other peroxisomal proteins (PTS2). There are separate receptors on the peroxisome for the two types....PTS1 and PTS2. While there are additional proteins required in the pathways, we will concentrate on just these two peroxisome signal receptors.
The cells of patients with peroxisomal disorders may contain only peroxisomal "ghosts" which is the empty membrane since the proteins cannot be internalized. Peroxisomal disorders are lethal but cells from affected individuals have been studied and complementation groups (CG) established. There are 11 complementation groups meaning there are a minimum of 11 proteins involved in the import pathways.
The peroxisomal disorders have been placed in three clinical groups: Group C have peroxisomes but A and B do not have peroxisomes but only "ghosts." CG refers to complementation groups.
Group A includes Zellweger Syndrome (ZS), infantile Refsum disease, neonatal adrenoleukodystrophy and hyperpipecolic acidemia, all characterized by severe neurological and hepatic dysfunction, craniofacial abnormalities and hypotonia, leading inevitably to early death. Patients accumulate phytanic acid (from plants) and very long chain fatty acids (VLCFA) in the blood and are deficient in the synthesis of plasmalogens, a class of phospholipids constituting up to 20% of our phospholipids.
Group B includes rhizomelic chondrodysplasia punctata (RCDP) These patients have severe growth defects, rhizomelia, cataracts, epiphyseal calcifications and ichthyosis. These patients accumulate higher levels of phytanic acid than group A patients, and have normal levels of VLCFA but lack plasmalogens.
Group C disorders involve the loss of only a single peroxisomal enzyme and the phenotype varies depending on which enzyme is deficient.
Group A and B diseases affect the localization of multiple peroxisomal matrix proteins and the medical prognosis for these affected individuals is bleak, with little chance of survival beyond the age of ten, particularly for the Group A disorders.
Group A patients fall into CG1-CG4 and CG6-CG10. CG2 have mutations in the gene, PEX5 which codes for the PTS1 receptor. The other complementation groups correspond to other proteins involved in both import pathways.
Group B patients (CG-11) all have a mutation in the PEX7 gene which codes for the peroxisomal PTS2 receptor.
Pharmacogenetic disorders
Pharmacogenetics is a term used to describe the study of genetic disorders that are induced by medications or other environmental factors. Some of these disorders would go undetected unless the individual with the "susceptibility" genotype encountered the drug, the food or environmental pollutant that causes a problem. The question is why some drugs work well in some patients but not in others. Importantly, 60-80% of variations in drug response may be genetic. Pharmacogenetic research studies the variability in drug response due to heredity. Research in this area is recent, wide-open, and very important. Adverse drug reactions are frequent and sometimes fatal and research in this field should help in reducing morbidity and mortality caused by prescription drugs. Studies in this field are leading to personalizing treatments to match the individual.
Genetic variation occurs at many levels: drug absorption, distribution, metabolism, receptors, and elimination. Single-nucleotide polymorphisms (SNPs) are being identified as a consequence of the Human Genome Project. These SNPs may change protein structure and function (missense), the level of protein expression, or RNA splicing. SNPs found in introns that can influence mRNA stability. SNPs have been shown to later drug uptake, excretion, and efficacy. Clinical studies on allergic disease have centered around SNPs that influence the drug target to alter efficacy. However, it is highly likely that a combination of SNPs (haplotypes) are more likely to influence function. There are wide variations that occur in SNPs in people of different ethnic groups and even between different groups within the same country. Studying these differences will explain the differences in drug efficacy and also differences in side effects.
Pharmacogenomics is a newly coined term which refers to new drug development based on our increasing knowledge of the human genome.....designer drugs. Common or clinically important pharmacogenetic variants are described below. Most of them are variations in drug metabolizing enzyme genes (or receptors) and drug transporter genes.
1. Glucose-6-phosphate dehydrogenase deficiency (G6PD) is an XR trait with more than 40 different mutations known. G6PD deficiency impairs the ability of the erythrocyte to form NADPH, a coenzyme used in reduction reactions in cells. The deficiency results in hemolysis in the erythrocyte when certain drugs, infections, fava beans cause oxidant stress. G6PD can be a cause of neonatal jaundice, most often in male babies. Hemolysis and jaundice occurs within 2-3 days of the drug dose (antimalarials, sulfonamides, nitrofurans,) G6PD is historically common in the Mediterranean region, where hemolytic anemia occurred after eating fava beans or Infection, rather than drug exposure which is now a common trigger of hemolysis. The diagnosis is made by a spectrophotometric assay to measure enzyme activity. Females who were heterozygous for the A and B alleles were the source of cells used initially for the study and proof of Lyonization. When their cells were grown in culture, it could be shown that they had two populations of cells, some of which made the A (electophoretically "fast") form and other cells, only the B. The mutant gene is uncommon in Europeans but relatively common in Africans and Mediterraneans.
2. Slow acetylator polymorphism (N-acetyl transferase). Both slow and rapid acetylator mutations exist and several different mutations are known. Drugs that cause problems are isoniazid (T.B. medication), hydralazine, procainamide, etc. Antinuclear antibodies are common within several months of starting procainamide (cardiac depressant). There is research now in progress to study heterocyclic amine carcinogens found in cooked meat.and the involvement of these mutations. Slow acetylators are prone to peripheral neuropathy on conventional doses of isoniazid. Rapid acetylators require larger does of medication and slow actylators less. Levels of the drugs in the body should be monitored. The diagnosis can be made by measuring the ratio of unmetabolized to metabolized forms of test drug in urine. Drug Reaction Testing
3. Debrisoquin poor metabolizer polymorphism (AHH aryl hydrocarbon hydroxylase is a P450 protein). These are enzymes of the smooth endoplasmic reticulum which are involved in the detoxification of exogenous substances. Both slow and rapid polymorphisms exist and several different mutations have been identified. Mutations have been found in 5% of the population. There is evidence that specific mutations are associated with lung cancer in smokers. Mutations affect the metabolism of some hypertension medications, -adrenergic blockers, some tricyclic antidepressants and antiarrhythmics. Megadosing may be needed for the ultra rapid metabolizer. The condition is diagnosed by measuring the ratio of un metabolized to metabolized forms of test drug in urine
4. Pseudo cholinesterase deficiency (succinyl choline sensitivity) occurs in approximately 1/3500 Caucasians. The disorder associated with the enzyme deficiency is precipitated by muscle relaxants such as halothane and succinyl choline. The disorder can cause problems in shock therapy patients. The resulting muscle paralysis and apnea can last hours rather than several minutes after drug dose. The condition is diagnosed by the Dibucaine number = % inhibition of hydrolysis of substrate in presence of 10 µM dibucaine.
5. Malignant hyperthermia (ryanodine receptor) Several different mutations have been identified with both locus and allelic heterogeneity. The disorder is triggered by inhalation of anesthetics (chloroform, ether) and succinyl choline. The condition is also found in pigs who experience the condition due to acute stress prior to slaughter. The result is that their meat is inedible due to the release of some (bad tasting) substance. Therefore, there is a need to identify the pigs with the condition ahead of time so they do not breed. The pathophysiology involves the abnormal regulation of intracellular Ca2+. Tachycardia and other arrhythmias occur early; acidosis, muscle rigidity and temperatures as high as 43o C occur later. The diagnosis is made using a muscle biopsy with a measurement of contraction of the living muscle during exposure to halothane or caffeine. The treatment is the use of Dantrolene. Malignant hyperthermia home page
6. Another disorder precipitated by environmental agents is alpha1-antitrypsin deficiency (14q32.1). This protein is the main plasma protease inhibitor (PI). It is especially needed to inhibit the action of elastase in the bronchial system of the lung. The clinically most important mutants are the Z and S alleles. The Z mutant protein aggregates in the ER-Golgi and the S mutant protein is degraded and the disease is due to the deficiency of the PI which then results in lung disease (emphysema). ZZ homozygotes also have liver disease. There is evidence that the carriers may also be affected and should, like the homozygotes, ZZ or SS, or compound heterozygote, ZS, avoid dust, smoke or smoking . Diagnosis can be made by AAT levels (IEF) or ASO DNA analysis.
7. Additional ecogenetic disorders and the environmental factors include: FH and cholesterol, lactase deficiency and milk, atypical alcohol dehydrogenase (ADH) and alcohol, and hemochromatosis and food containing iron.
Clinically, hereditary hemochromatosis affects 1/200 people. It manifests as cirrhosis of the liver, pancreatic damage leading to diabetes, hormonal abnormalities, depression, hypermelanotic (bronze, dark) pigmentation of the skin, chronic fatigue, joint pain, loss of libido, heart problems, abdominal pain, and heart failure, all due to iron overload. The disorder is treated by phlebotomy. Before menopause, most women with hemochromatosis lose enough blood during their menstrual periods to avoid dangerous iron buildup. After menopause, iron begins to accumulate and some of the symptoms such as fatigue, joint pain, depression, diabetes, and heart and liver problems may be mistaken for signs of menopause or aging. This is why the disorder can go unrecognized and undiagnosed for evens, even though it is one of the most common genetic disorder in the United States. The body needs only small amounts of iron; we absorb about 10% of the iron we consume.
Most of the time HH results from a mutation in the HFE gene, which helps regulate iron absorption by the intestines. The most common mutation, called C282Y, is involved in about 90% of cases and 83% of those affected are homozygous for this allele. Homozygotes for this mutation absorb too much iron, and up to 40% will develop the disorder. Symptoms are more common for those who consume large amounts of iron-rich foods, iron supplements, or alcohol which increases iron absorption. The HLE gene is closely linked to the HLA genes on chromosome 6 (6p21.3) and is homologous to them. Early diagnosis of HH and therapy can prevent organ damage. The most common mutation that causes hemochromatosis is a missense mutation in a highly conserved residue involved in the intra molecular disulfide bridging of MHC class I proteins. The mutation disrupts the structure and function of the protein. A G-to-A transition at nucleotide 845 results in a cys282-to-tyr (C282Y) substitution.
Carriers may have slightly increased iron absorption but seldom develop the classic symptoms unless they drink excessively or have another liver problem such as hepatitis C. The carrier frequency is 6.4%. The geographic distribution of the allele suggests a Celtic origin. Another common mutation is H63D where a histidine is replaced by an aspartic acid (his63-to-asp). While this mutation alone does not cause HH, it is often found with C282Y in compound heterozygotes with HH.
Common blood tests include transferrin saturation (TS), which measures the percentage of iron bound to the protein that carries it into the blood-stream, and serum ferritin, which provides a rough gauge of iron stores in tissues. It these are abnormally high, a blood test to detect the HFE mutation can confirm the diagnosis. In women, a TS of 50% or more and a serum ferritin concentration os 200 ng/ml or more (300 ng/ml if postmenopausal) suggest iron overload. The CDC (Center for Disease Control) recommends testing of close relatives of people with HH and those with otherwise unexplained symptoms common to HH. Treatment is repeated phlebotomy.
Some experts regard HH as the perfect candidate for genetic screening. The disease is common and potentially deadly, and permanent damage can be prevented if the diagnosis is made before symptoms appear. While DNA tests are available for the most common mutations, they are not perfect. About 20% of people with biopsy-proven illness do not have the recognized mutation. Of those with the mutation, not all will develop symptoms. And although their condition is highly treatable, people with hemochromatosis have reported difficulties obtaining medical and life insurance.
The National Heart, Lung, and Blood Institute is conducting a 100,000-person study (to be completed in 2005) to determine the prevalence of hemochromatosis. It will also search for other genetic variants that might promote iron accumulation and evaluate the ethical, legal, and social implications of genetic screening. American Hemochromatosis Society
Addendum: Some Mendelian (Single Gene) Traits....AD, AR, XR
We have covered some of these previously and will cover others in future lectures. However, this is your cue to look them up in your textbooks and learn about their etiologies.
Mendel considered the father of genetics. He became an Abbey of his monastery for reasons after his work was not recognized for its amazing insights into heredity. He did his genetic crosses around 1865 and was rediscovered by three independent researchers around the turn of the century. Mendel did not coin the word "genes", Bateson did, Mendel used the term "factors." He also did not know about chromosomes nor did he know about linkage. All of the traits he studied were either on different chromosomes or so far apart on the same chromosome that they assorted independently. He may have found his law of independent assortment did not work for all two factor crosses (if they were linked) and gave up and became an administrator. Bateson was the first one to describe linkage. In human genetics we rarely work with more than one pair of genes at a time so we, like Mendel, need not concern ourselves with linkage very often.
Achondroplasia is an AD trait that is fully penetrant. It is the most frequent form of short-limb dwarfism. About 7/8 of cases are the result of new mutation, there being a considerable reduction of effective reproductive fitness. There is a paternal age effect. Gonadal mosaicism (or spermatogonial mutation) is a possible explanation for the occasional report of affected sibs from normal parents. Affected individuals have rhizomelic shortening of the limbs, a characteristic facies with frontal bossing and mid-face hypoplasia, exaggerated lumbar lordosis, limitation of elbow extension, genu varum (bow legs), and trident hand. The mutation is in a gene coding for a fibroblast growth factor receptor (FGFR-3) located on chromosome 4 at p16.3. People with AD traits if they are rare are heterozygous and rarely does one get to see the homozygous phenotype. However, since individuals with achondroplasia meet in organizations such as Little People of America, there have been mating. When this occurs about 25% (overall) of the offspring are severely affected and often die in utero. Therefore, the trait is not a true AD since two doses of the mutant gene is obviously not equal in severity to one. 25% of the offspring of a mating between heterozygotes will be of normal stature and 50% will have achondroplasia like their parents. Usually the matings are between heterozygotes and homozygous normal individuals, in which case the probability of having a child with normal stature or with achondroplasia is equal (50%).
Crouzon syndrome is an AD craniofacial dysostosis also due to a mutation in a fibroblast growth factor receptor (FGFR-2) on chromosome 10. There is craniosynostosis, the eyes have shallow orbits, there is proptosis, hypertelorism, parrot-beaked nose, short upper lip, hypoplastic maxilla and mandibular prognathism.
Abraham Lincoln is believed to have had the AD Marfan syndrome but some question this. However, Paganini, the great violinist is believed by many to have had it. And, of course, the great athlete, Flo Hyman, died of the consequences of Marfan. This disorder is caused by a defect in fibrillin a connective tissue component. It is characterized by above average and disproportionate height, arachnodactyly, dislocation of the lens, joint laxity, striae, and scoliosis or kyphosis. It is sometimes treated with propanolol in an attempt to prevent aneurysms. Some basketball players with Marfan syndrome have died from it. Pregnant women with the syndrome need special care due to the stress put on the body.
Treacher Collins is an AD with variable expressivity. You saw a picture of a father and son. The son was much more severely affected and the father who only showed mild expression of the disorder was only diagnosed after the birth of his son. The phenotype can include a reduced mandible, malformed ears, deafness, cleft palate and mental handicap. The gene (TCOF1) is at 5q32-q33.1 and codes for a protein of unknown function called TREACLE.
Neurofibromatosis (NF1) is caused by the inheritance of a mutant tumor suppressor gene and the subsequent loss of heterozygosity for the gene in the affected tissues. The gene product is called neurofibromin. NF1 shows extreme variation in expression since the neurofibromas show up in a wide variety of locations in the body. If they are present in the brain they can cause mild to severe mental retardation. One manifestation is the presence of multiple café au lait spots on the body especially in the axillary region. The gene is extremely large and many cases are due to new mutations. Since mutations are known to occur at many sites within the gene, DNA analysis is difficult and instead a test is done to look for the truncated protein which is produced by the mutant gene (see handout).
Huntington disease is a true AD. It is caused by a TNR in the huntingtin gene. The function of the protein is unknown but it is widely expressed cytoplasmically. The mutant protein has a long stretch of glutamine residues which causes the protein to aggregate with other proteins. The folksinger, Woody Gurthrie, had the disorder and his son, Arlo, has a 50% risk of having inherited the mutation. Last seen, he was okay at about 50 years of age. The gene is found in a relatively high frequency with a small community in Venezuela. This is an example of a Founder effect where the gene was brought into the community by a European sailor who had many progeny. Since the community is small, there is a higher level of inbreeding and thus there are homozygotes known for the disorder but they do not manifest the disorder any differently than the heterozygotes. Thus the disorder is a "true" dominant. DNA testing for the trinucleotide expansion is available. People can now know ahead of time if they carry the disorder. This has lead to interesting ethical dilemmas when one member of a family wants to know their status and the parent (or child) does not. Also, there is some question as to the age at which a person can make an informed choice to know. Anyway, testing can be a problem in these families.
(Cornelia) de Lange, thanatophoric dwarfism and progeria are examples of new sporadic AD mutations which are also genetic lethals. TD is a perinatal lethal. Cornelia de Lange syndrome includes severe mental retardation, synophrys, hirsutism and limb deformaties.
Progeria results in premature aging with most affected persons dying in their teens. It does not affect mental abilities.
Freckles show an autosomal dominant pattern of inheritance but there is evidence of gene dosage effect as for achondroplasia and familial hypercholesterolemia. In other words, the homozygote may have more freckles than a heterozygote. By the way, freckles are found in all ethnic groups.
Retinoblastoma, like NF1, is caused by the inheritance of a mutation in a tumor suppressor gene. The cancer is caused by a loss of heterozygosity and occurs in early childhood. The retinoblastoma is usually bilateral and of early onset if inherited.
Epidermolysis bullosa results in blisters following minor trauma to the skin. EB is a group of hereditary diseases of the skin and both AD and AR patterns have been proposed. In the simplex (heals without scarring) type, epidermal basal cells are fragile and mutations of genes encoding keratin intermediate filament proteins underlie that fragility. In the dystrophic types, the causative mutations (both AD and AR) appear to be in the gene encoding type VII collagen, which is the major component of anchoring fibrils. These findings provide evidence that the cytoskeletal intermediate filament network is necessary for mechanical stability and that anchoring fibrils anchor the epidermis to the underlying dermis.
Postaxial polydactyly type A1 (PAP-A) is an AD trait with wide variation in how many hands and feet show the extra digit. It is also incompletely penetrant and an (apparent) unaffected person may have an affected child. This form of polydactyly is about 10 times more frequent in blacks than in Caucasians. From the study of various pedigrees, it is suggested that 2 phenotypically and possibly genetically different varieties exist. In type A, the extra digit is rather well formed and articulates with the fifth finger or an extra metacarpal. This type is inherited as an AD with marked penetrance. In type B (pedunculated postminimi), the extra digit is not well formed and is frequently in the form of a skin tag. The genetics of this type is more complicated. Using a 5 generation Indian family in which 15 individuals were affected a genome wide search showed linkage to markers on chromosome 7 and a candidate gene was identified. In a large Turkish family, the trait was linked to DNA markers on 13q. This was referred to as type A2 and the form in the Indian family as A1.
Phenylketonuria (PKU) is an AR trait. It is called an inborn error of metabolism because it is due to the malfunctioning of an enzyme. Classic PKU is due to mutations in the gene for phenylalanine hydroxylase (PAH) which converts phenylalanine to tyrosine. It is common enough (1/104 live white births) and serious enough (severe mental retardation) to be included in the newborn screening programs of all states. The lack of PAH causes toxic levels of the amino acid, phenylalanine, to build up in the blood which then damages the brain. A diet restricted in the amount of phenylalanine can prevent the serious mental retardation. The newborn screening is called the Guthrie test. A drop of blood from a baby who has had some food (milk) is dried on a small filter paper disk. The disk is placed on an agar plate containing B. subtilis and thienylalanine. If there are high levels of phenylalanine in the baby's blood, the growth of the bacteria will not be inhibited by the thienylalanine and you will observe bacterial growth around the test disk. A positive Guthrie test is followed by a quantitative assay of phenylalanine and tyrosine in the baby's blood before a definitive diagnosis of PKU is made. There is allelic heterogeneity due to different mutations in the phenylalanine hydroxylase (PAH) gene. Four common alleles account for the majority of cases among northern Europeans. However, there are other alleles in other ethnic groups. The frequency of the different alleles varies with different ethnic groups and many people affected with PKU are compound heterozygotes. Some of the mutations result in milder phenotypes. Mutations in genes at other loci can also cause hyper phenylalanemia. These genes are involved in the synthesis of tetrahydrobiopterin (BH4) or in its regeneration. BH4 is a cofactor in the reaction which converts phenylalanine to tyrosine which is carried out by PAH. One such gene is one that codes for the enzyme dihydropteridine reductase (DHPR) which reduces quininoid dihydrobioterin (qBH2) to tetrahydrobiopterin (BH4). Treatment is very different for these patients. Maternal PKU leads to severe MR in the fetus due to the high levels of phenylalanine in the blood. Therefore, females must remain on the restricted diet if they are to have normal children.
Tay Sachs Disease (TSD) is an AR inborn error of metabolism. It belongs to a family of genetic disorders known as the lysosomal storage diseases. Lysosome which are found in all cells of the body contain a variety of acid hydrolases which break down a variety of substrates which are large molecules. If any one of the hydrolytic enzymes is missing, there is a build up of the substrate in the lysosome. The organs affected depend on which enzyme is missing and where its substrate is primarily found. The enzyme missing in Tay Sachs disease is hexoseaminidase A, an enzyme that cleaves monosaccharides from oligosaccharides which are part of a lipid known as a ganglioside. These gangliosides are found primarily in the CNS and the lysosomes become engorged with these lipids.The accumulation kills the cells in the brain and CNS and the result is death in infancy. There is no known cure. Effective screening programs have been implemented among Ashkenazi Jewish communities where the carrier frequency is 1/27 as opposed to about 1/300 among other ethnic groups. Prenatal detection is also possible. The gene has a high incidence among some French Canadians as well. The higher incidence in some ethnic groups is due to founder effects. The higher incidence may be due to some as yet undiscovered heterozygote advantage. As in all cases we study, there is alleleic heterogeneity and locus heterogeneity. Some alleles which only reduce the enzyme activity result in a later onset.
Xeroderma pigmentosum (XP) is a disorder with locus heterogeneity. The defects are in the DNA excision repair pathway for UV damage. UV causes a TT dimer to form which must be identified and excised by an endonuclease, a section then removed by an exonuclease, a DNA polymerase and finally a ligase. This presents us with at least 4 required enzymes and each enzyme can be a multimer. Complementation tests have shown at least 9 complementation groups. The interpretation is that there are at least 9 genes involved in XP. Some forms affect the CNS and some do not. Skin cancer is initiated by UV mutations and progresses to cancer when these cells are not triggered by normal processes to go into apotosis.
Albinism exhibits locus and allelic heterogeneity. Albinism is often divided into tryrosinase positive and tyrosinase negative types. In oculocutaneous albinism known as OCA type IA (classic OCA) tyrosinase activity is absent. In type IB (yellow mutant) there is a partial deficiency of tyrosinase activity. There is some clustering of missense mutations in tyrosinase one such site is the binding site for copper, a cofactor for the enzyme. OCA type II individuals make a small amount of pigment and have light yellow hair. Both type I and type II lack visual pigmentation but type II has milder visual defects than type I. In type II, the major gene is referred to as the P gene which is thought to be involved in tyrosine transport into the cell. P refers to the pink-eyes of the phenotype. A temperature sensitive albinism has been reported in a few individuals who have white hair in warmer areas of the body and darker hair in the cooler extremities. This is the same as albinism in Siamese cats and Himalayan rabbits. Females heterozygous for an XR form of albinism show pigmented and non-pigmented patches in the in retina due to the mosaicism produced by Lyonization.
Lesch Nyhan syndrome is due to the deficiency of an XR enzyme, hypoxanthine phosphoribosyltransferase (HPRT). The extreme phenotype of self-mutilation, mental retardation, spasticity and high levels of uric acid leading to gout is associated with the lack of the enzyme. This enzyme is involved in purine metabolism which can affect levels of neurotransmitter among other things. There is allelic heterogeneity and other mutations in the gene lead to milder phenotypes such as gouty arthritis.
Fragile X is another X linked trait we have already studied as an example of a TNR disorder. Both males and females can be affected. Approximately 50% of the male offspring of a carrier mother and 30% of her daughters will be affected. Non manifesting carrier males can pass the premutation to their daughters who have a high risk for having affected offspring since there is high probability of expansion in premutation females than premutation males. The gene, FMR-1, is expressed in the CNS but its exact function is not known. The TNR (CGG repeats) occurs in the promoter region of the gene and when greatly expanded leads to methylation and inactivation of the gene.
Duchenne Muscular Dystrophy (DMD) is caused by mutations, mostly deletions, in the unusually large gene for a large rod-like protein called dystrophin. Dystrophin has some homology to spectrin, an actin-binding-protein and has been found in smooth, skeletal and cardiac muscle. It is localized to the cell membrane, confirming its role in the cytoskeleton. DMD is due to the complete absence of dystrophin usually caused by large deletions. Another disorder, Becker Muscular Dystrophy (BMD) is also due to mutations in the dystrophin gene. However, BMD is milder than DMD because the mutations result in either less dystrophin or an abnormal dystrophin instead of no dystrophin.
Another XR disorder we have already discussed is androgen insensitivity/testicular feminization (AIS/Tfm). This disorder is both sex linked AND sex limited. It is sex linked because its gene, the androgen receptor, is on the X chromosome. It is sex-limited because it only manifests in XY fetuses. There are a variety of mutations in the androgen receptor (allelic heterogeneity) and there are also a wide variety of phenotypes produced by the different mutations in this gene. The phenotypes range from an phenotypically normal male who is merely infertile to a person with the outward appearance of a female but who has testes and no female internal plumbing.
