BIO 442 MENU
syllabus 
1 - genome
2 - mutate
3 -cell cycle
4 - karyotype
5 - chromoabn
6 -sex-determ
7 -prenatal
8 - mendelian
9 - complex
10 - non-trad
11 - clinical
12 - newborn
13 - teratog
14 - linkage
15 - DNA prof
16 - quanti
17 - links
18 - quizzes
(full title of lecture appears in status bar on the top or at the bottom of your window)

Biology 442 - Human Genetics
Chromosome Abnormalities: Autosomes and Sex Chromosomes
Chromosome aberrations are classified as one of two types: numerical or structural. Numerical changes are to two types: polyploidy with changes in the number of sets of chromosomes (polyploidy) and aneuploidy with changes in the number of individual chromosomes (e.g., trisomies and monosomies). Structural changes involve the loss or gain of portions of chromosomes. The resulting patient may be said to have "partial monosomy" or "partial trisomy."
Numerical Chromosome Abnormalities
It is estimated that 10% of sperm and 50% of eggs contain abnormal chromosomes (both numerical and structural). Several studies have shown that more than 50% of first trimester spontaneous abortions (SABs) are due to chromosome abnormalities. Most are due to trisomies with trisomy 16 being the most common autosomal trisomy. Monosomy X (Turner Syndrome) is as common as trisomy 16. Triploidy (3n), which is mostly due to dispermy, is the next most common chromosome abnormality in SABs. Only 5% of stillborn have chromosome abnormalities and only 0.5% of newborn have chromosome abnormalities.
Almost all chromosome trisomies have been identified in abortuses but some are lethal prior to implantation and, therefore, before the detection of a pregnancy by standard techniques. Most trisomies are due to maternal non disjunction in meiosis I; monosomy X is frequently a consequence of non disjunction in male meiosis I. Tetraploidy (4n), is due to a post zygotic non disjunction in mitosis. Hydatiform moles are due to an anucleate egg being fertilized by either one sperm which undergoes endo reduplication to produce a totally homozygous condition or due to dispermy. In either case, the contribution is totally the male parent's chromosomes and is not compatible with life. The grape like fleshy cluster that forms is primarily extra embryonic tissue. Teratomas, on the other hand, have totally maternally derived chromosomes and are composed of immature embryonal elements derived from all 3 germ layers. They are tumors and can become malignant if not removed. The moles and teratomas have the requisite 46 chromosomes but in each case are derived solely from either the female or male. These are examples of genetic imprinting a phenomenon that occurs during meiosis and which results in the male and female genetic contributions to the zygote not being the same.
Aneuploidy

Trisomy 21 (Down Syndrome)
Trisomy 18 (Edward Syndrome)
Triploidy
Frequency and Distribution of Chromosome Abnormalities in Different Categories of Clinically Recognizable Human Pregnancies
FREQUENCY OF ABNORMALITY (%)
| Chromosome abnormality |
Spontaneous abortion |
Stillbirths | Live births | Probability of survival to term |
| All | 50 | 5 | 0.5 | 5 |
| Trisomy 16 | 7.5 | - | - | 0 |
| Trisomy 13,18,21 | 4.5 | 2.7 | 0.14 | 15 |
| XXX,XXY,XYY | 0.3 | 0.4 | 0.15 | 75 |
| All other trisomies | 13.8 | 0.9 | - | 0 |
| 45,X | 8.7 | 0.1 | 0.01 | 1 |
| Triploidy 3n | 6.4 | 0.2 | - | 0 |
| Tetraploidy 4n | 2.4 | - | - | 0 |
| Structural Abnormalities | 2.0 | 0.8 | 0.3 | 45 |
Importance of Errors at Different Stages as Determined by Studies of Spontaneous Abortions
STAGE OF ERROR (%)
| Abnormality | Female meiosis I |
Female meiosis II |
Male meiosis I |
Male meiosis II |
Fertilization Polyspermy |
Early cleavage division |
| Trisomies | 75-95 | 5-25 | 0 | 0 | little, if any | |
| 45,X | 20 | 80 | 0 | |||
| 3n | ~25 | 0-25% | 50-75 | 0 | ||
| 4n | 0 | 0 | 0 | 0 | 0 | ~100% |
| Hydatiform mole, 2n, all paternal |
Failure of oögenesis leading to anucleate egg |
Anucleate egg fertilized by single sperm which undergoes endo reduplication or, more rarely, fertilization by two sperm |
| Teratoma, 2n, all maternal |
Failure of oögenesis leading to diploid egg which begins to differentiate in the ovary (involves all 3 germ layers) |
Chromosome Aneuploidies in Newborns
Down Syndrome
Down syndrome (DS) is the most common (1/900 = 0.11%) viable autosomal trisomy in live borns. Chromosome 21 is shorter than chromosome 22 but was misnamed so long ago that they left the number as 21. Therefore, this trisomy is tolerated in triplicate probably because it represents the least genetic imbalance of the trisomies. The frequency of Down Syndrome in spontaneous abortions (SABs) is 3%. As is true for all autosomal trisomies, it is most commonly associated with advanced maternal age. If the parents have informative DNA marker alleles (do not have identical alleles) the parent of origin of the extra 21 can be determined and these markers can also tell whether the non disjunction occurred in meiosis I or II (see figure below).
This trisomy 21 child received an extra maternal chromosome 21 as a consequence of non disjunction in Meiosis I
Down syndrome children have multiple anomalies. All are mentally retarded and hypotonic. They usually have heart defects, GI tract problems, respiratory illnesses, early Alzheimer disease, and leukemia. Female DS are fertile and have a 50% chance of having a DS child. The DS male is sterile. A phenotype map of chromosome 21 can be constructed which shows how children with varying amounts of triplicate copies of regions of chromosome 21 have only some of the features of the full trisomy 21 Down syndrome (see figure below).
Down syndrome can be due to a straight trisomy, a translocation of 21 to any of the D or G group chromosomes, a mosaic condition where there are some normal and some trisomic cells, or a duplication of part of the q arm of 21. Inherited Down Syndrome usually means that a parent carries a balanced Robertsonian translocation (centric fusion) between 21 and a D or G group chromosome. These families show, in addition to increased numbers of Down syndrome children, an increased number of SABs due to other imbalanced chromosome complements in their gametes. Occasionally a family or an individual will have multiple trisomy cases. These might be explained by an inherited tendency for greater non disjunction (problem with kinetochore? spindle?) and they are given a recurrence risk of 1%.
95% of Down syndrome patients are trisomy 21 and 4% are unbalanced translocations of the 21 with a D or G group chromosome. A very few Down syndrome patients are mosaics. In general, mosaic Down syndrome cases are better off physically and mentally depending when in mitosis of the embryo the non disjunctional event occurred. (Even you may have trisomy 21 in your big toe.) Translocation Downs may arise de novo in the egg (usually) or sperm or it may be inherited from a parent who has the balanced translocation. In general, male carriers of a balanced translocation, 45,XY,t(14;21) have a lower recurrence risk (< 5%) than a female carrier, 45,XX,t(14;21) (10 - 15%). (Does the sperm with less chromosomal material swim upstream faster then the one with the extra 21?) Parents of Down syndrome children with straight trisomies are given a recurrence risk of 1% based on empiric observation. (Some families may be at higher risk for non disjunction than others.) Trisomy 13 (Patau syndrome) and trisomy 18 (Edward Syndrome) are two other autosomal aneuploidies that are found in live born infants. These infants, unless they are mosaics, usually die within a few days or months.
Down syndrome (trisomy 21) and Turner syndrome (45,X)are both called aneuploidies. However, Down syndrome is an autosomal trisomy and Turner is a sex chromosome monosomy. Turner Syndrome is the only viable human monosomy. It is described later in this page.
Uniparental disomy
Uniparental disomy is the inheritance of two homologous chromosomes (autosomes or X) from one parent. This can occur in a normal diploid fetus or a trisomy fetus. Isodisomy is the result of non disjunction either in Meiosis II when chromatids normally separate or a post zygotic mitosis. With isodisomy the individual inherits two identical (except for crossing over) homologs from one parent and is homozygous of many or most of the genes on those chromosomes. This can lead to deleterious effects. Heterodisomy refers to the inheritance of two different homologs from the same parent. It is the result of a meiosis I non disjunction in one parent. Robertsonian translocations occur between the D and G group chromosomes. When they involve the same chromosome this often results in UPD. An autosomal trisomy will by definition have either heterodisomy or isodisomy.
A fetus may start out diploid and become trisomic through a mitotic error. The converse can also happen, an embryo can begin trisomic and lose the extra chromosome and become diploid. In both cases the embryo will have uniparental disomy (UPD), two homologs from one parent and depending on how the chromosomes segregated the embryo, it may have cells that are heterodisomic (unlike) or isodisomic (like) for the chromosome in question. Uniparental disomy can have consequences for the fetus. If the fetus is diploid and has UPD and there are genes on the chromosome that are "imprinted" differently in the male and female parent, the fetus may have a disorder such as Prader Willi or Angelman syndrome. If the fetus is trisomic and there is isodisomy the consequences may also be a double dose of whatever "bad" genes were present on the "double dose" chromosome.
Transient leukemia (TL) is present in approximately 10% of Down Syndrome (DS) newborns. These infants do not show a maternal age effect which is associated with meiosis I errors. In these cases, it is likely that the non disjunction was in meiosis II or mitosis in the embryo and the result was isodisomy of one chromosome 21 and possibly a double dose of a gene predisposing them to leukemia. It is already known that DS children have a higher incidence of leukemia than normal children and that their siblings are also at an increased risk. All of this supports the existence of a susceptibility gene for leukemia on chromosome 21.
Paternal Contribution to Aneuploidy
The relationship of maternal age to chromosome aneuploidy is well known. Evidence for paternal contribution to aneuploidy has been conflicting. Recent studies have suggested that 10-30% of autosomal trisomies arise during paternal meiosis. Specifically, trisomy 18 is more commonly associated with advanced paternal age. Of the sex chromosome aneuploidies, 100% of XYY and 50% of XXY's are paternal in origin. At least 80% of Turner females have their mother's X chromosome and are therefore missing the father's X or Y. Uniparental disomy of chromosome 15 is correlated with advanced paternal age. Studies of sperm by Bosch et al (2002) using 4 color FISH probes for chromosomes 6, 21, X and Y showed a significant increase in the level of autosomal disomy and a smaller but significant increase in sex chromosome disomy with increasing male age. Triploidy is also increased with paternal age. Significant individual variation was observed among the study subjects.
Sex Chromosome Abnormalities
Aneuploidies involving the X and Y chromosomes are better tolerated than those involving the autosomes. This is because the Y chromosome contains no essential genes....except for maleness and male fertility and because only one X is active when more than one is present. (See later section on X inactivation).
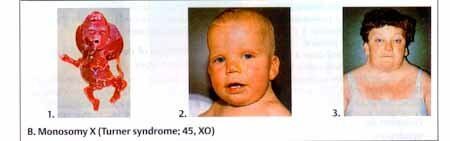
Turner Syndrome, 45,X
Turner Syndrome or monosomy X is the only known viable monosomy. It is reported to occur in 0.03 % newborns and 8.7% of SABs. It is one of the three most common chromosome abnormalities found in first trimester SABs. Since the condition is relatively benign in the live born Turner female, it is somewhat of a mystery as to why it causes early fetal death. Turner syndrome fetuses often have extensive edema. The edema in the neck and hands results in neck webbing and arched nails. The condition is not correlated with maternal age. Instead, non disjunction in male meiosis I accounts for 80% of cases. In general, abnormalities involving the sex chromosomes are better tolerated than autosomal abnormalities because only one X is active in the normal adult. Any extra X's are inactivated and the Y carries very little genetic information. Turner females are very short, are sterile due to gonadal dysgenesis, experience primary amenorrhea, have broad chests, and usually have heart defects and kidney malformation.
In 80.3% of Turner syndrome individuals the paternal sex chromosome is lost. Incidence is 1/2500 to 1/5000 live born females making its prevalence low compared to the other sex chromosome aneuploidies. 15/1000 clinically recognized pregnancies are 45,X and greater than 99% do not survive beyond 28 weeks gestation. Of those that do not survive: 1. Most are SABs during first trimester and consist of a chorionic and amniotic sac with a cord attached to a fragment of embryonic tissue or a small macerated embryo. 2. A small number are ruptured sacs without cord or fetal development. The single X in these cases is more likely to be of paternal origin. Others have reported that the Turner Syndrome females with the paternal X have slightly better verbal IQ scores and better social cognition. If either of these claims turn out to be true, it would be another example of genomic imprinting. 3. Some 45,X fetuses present later as second trimester abortion or stillbirths with fetal edema, hydrops, or nuchal mass. It is postulated that live born Turner Syndrome probably begin as euploid (46,XX or 46,XY) embryos.
Turner syndrome chromosome complements:
| 45,X | 50% |
| Approximately 50% of Turners are mosaic | |
| 46,X,i(Xq); 45,X/46,X,i(Xq) | 28% |
| 45,X/46,XX; 45,X/47,XXX | 13% |
| 45,X/46,XY | 5.5% |
| 45,X/46.X+mar (need to check for SRY) | 3% |
| One can also see 45,X/46,X,r(X) |
Although most Turner patients are infertile, there have been at least a dozen reports of fertility in the absence of any evidence of mosaicism. These women have increased risk of chromosomal errors and high incidence of fetal wastage--prenatal diagnosis is strongly recommended. It is prudent with any Turner syndrome patient or any prenatal diagnosis of Turner syndrome, to rule out mosaicism with Y chromosome material because of the increased risk for gonadal blastoma (gene responsible for gonadal blastoma is believed to be proximal to the centromere on the Yq). Turner syndrome patients should be referred to cardiology, urology, audiology, weight gain clinic, hypertension clinic, and endocrinology for growth hormone therapy and later for female hormone therapy.
50% of Turner females are 45, X. 26% have structural abnormalities: 17% iX, 2% Xp-; 7% rX. 20% are mosaic: 45,X/46XX; 45,X/abnormal X; or 45,X/47,XXX and 4% of mosaic cases were XY conceptuses who lost the Y in some cells and are: 45,X/46,XY. These females are virilized at birth and again at puberty and they have a 20% risk of malignancy of the dysgenic gonad. If Y chromosome material is found in a Turner female, the gonads should be removed.
There is a gene, SHOX, on the pseudoautosomal region of both X and Y. Turner females show haploinsufficiency for this gene and are, therefore, shorter than an XX female. In fact, the more sex chromosomes (X or Y) you have the taller you are! The Xq arm has genes for both ovarian development and maintenance. Turner females have oocytes during fetal life but they degenerate. It is believed that two functional X chromosomes are needed for normal ovarian development in fetal life. Interestingly, XO mice are fertile.
Turner females are diagnosed by their unusually short stature (less than 5 feet), webbing of the neck, heart problems (some of which can show up later in life), kidney malformation, and at the normal time of puberty, primary amenorrhea. Many Turner females are better at verbal skills than spatial skills. The 45,X is not correlated with maternal age and 80% contain the maternal X.
Noonan Syndrome is an autosomal dominant (AD) trait whose phenotype overlaps with Turner Syndrome (webbed neck, short, heart defect). However, both males and female are affected and they are fertile. Noonan syndrome has a normal male or female karyotype. The heart defect in Noonan is often pulmonary valve stenosis while in Turners it is coarctation of the aorta and atrial septal defect. These types of situations are the things one must be mindful of in diagnosis, prognosis, treatment, and counseling for recurrence risks.
Triple X Syndrome, 47,XXX.
Triple X has an incidence of approximately 1/1000 female births. 93.5% result from maternal non disjunction. 47,XXX females are tall (extra SHOX genes), are usually fertile but a significant number have urogenital problems including infertility. If fertile, there is an increased risk of chromosomal abnormalities. They have delays in language, neuromotor, and learning skills and have impaired communication and psychosocial adaptation. Increasing numbers of X chromosomes are correlated with mental handicap (XXXX, etc.). Triple X females are taller than their sisters. This is probably also a consequence of the SHOX genes on the X chromosome. Although most triple X females appear normal physically and are usually fertile, our clinic had a 47,XXX female patient who on ultrasound was found to have no uterus or fallopian tubes. On researching the literature we found other triple X females with Müllerian duct agenesis. This condition is often referred to as the Rokitansky sequence (see lectures on sexual differentiation).
Klinefelter male, 47,XXY.
Klinefelter Syndrome Web Site, Medical Klinefelter Page
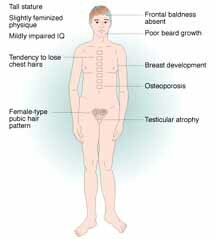
The Incidence of 47,XXY, is 1/1000 male births. The origin of the extra X chromosome is maternal 50% of the time and paternal 50% of the time. 72% of the cases are a meiotic I error. 97% of the fetuses survive to term. Klinefelter is the most common cause of hypogonadism, azoospermia or oligospermia. Most Klinefelter males are infertile, however, sexual function is normal but with decreased libido. Testosterone therapy is commonly necessary for maintenance of secondary sexual characteristics. XXY males have a feminized physique and pubic hair pattern, have a greater tendency to osteoporosis, are less apt to be bald, and gynecomastia in present in 1/3 of adult patients. Klinefelter males have autoimmune susceptibility, a high mortality rate for cerebrovascular disease (increased 6X over general population), a normal IQ although often less than sibs, and their reading skills are often poor.


A "Klinefelter" calico cat provided some of the first evidence that the Y chromosome determines maleness in mammals. Normally, Calico cats are female since the genes for black and gold are alleles and are carried on the X chromosome. The pattern of circles of black and gold are examples of the random inactivation of one of the female's two X chromosomes. When a male calico cat was found it was shown to have an XXY chromosome constitution. Like its human counterpart, it was sterile.
Klinefelter Variants:
| 48,XXXY | More severe clinical presentation than 47,XXY. Usually are mentally retarded |
| 49,XXXXY | Moderate to severe mental retardation; marked hypogonadism; skeletal abnormalities; congenital heart disease |
| 48,XXYY | Taller but much like 47,XXY; phenotypic overlap with XYY |
47,XYY male.
The birth incidence of 47,XYY is about 1/1000. There is no parental age effect. 100% are paternal in origin and are the result of a paternal meiosis II non disjunction. They are not mentally retarded but their IQ is generally lower than their sibs. They are taller than their male sibs (again extra SHOX genes on the extra Y). While there is no real phenotype, these males show a higher incidence of distractibility and have impulse control problems. They may have increased risk for chromosomal abnormalities in their offspring.
X Chromosome Inactivation
The normal female has two X chromosomes, yet the possession of only a single X is sufficient for normality in the 46, XY male. The second X is not all needed and in humans (and all mammals) dosage compensation of X-linked genes is accomplished by the random transcriptional silencing of one of the two X chromosomes in the female during early development (about the second week following conception). This is called X inactivation or lyonization (named for Dr. Mary Lyon). The random inactivation is clonal, all descendent cells inactivate the same X (either the paternal or the maternal X). This mechanism is required to ensure equivalent levels of gene expression from the sex chromosomes.
Early events of X inactivation are under the control of the X-chromosome-inactivation center (Xic). Inactivation requires a specific cis acting signal, namely the RNA product (X inactivation-specific transcript) of the XIST gene (Xq13.2) found in the XIC (X inactivation center). This gene is active only on the inactive X chromosome. At the time of inactivation, the XIST RNA functions in cis (cis means on the same chromosome) to spread an inactivating signal in both directions, up and down the inactive X chromosome on which it resides. The action of the signal is believed to result in methylation of DNA cytosine residues in the 5' CpG islands of the silenced genes. The result is referred to as facultative heterochromatin. The XIST gene has no ORF (origin of reading frame) and its product is a repetitive RNA sequence. Initiation of X inactivation involves a step in which the number of X chromosomes in the cell is counted relative to the cell ploidy so that only a single X chromosome is functional in each diploid cell. Interesting research is underway to determine the way in which cells selectively silence one X but not the other in the same cell, how the X that is silenced is chosen, how the number of X's in a cell is counted, and the silencing is accomplished rapidly and efficiently during early development.
It is important to note at this juncture that CpG methylation (m5C) is a common mechanism in the down regulation of gene expression, it serves to silence genes. There are several different DNA methyl transferases (DNMT) or methylases. Methylation is required for normal development and the absence of methylation in genetically engineered mice is lethal. There is hyper and hypomethylation involved several known disorders including cancer, Prader Willi and Angelman syndromes, Beckwith Wiedeman and lupus. Abnormal methylation has been implicated in in vitro fertilization problems. Loss of heterozygosity (LOH) can be due to methylation of a normal tumor suppressor gene when its allele is mutated or deleted. Cancer then is the result.
Not all genes on the "inactive" X are silenced. This is obvious when one compares the condition of 46,XX and 45, X, Turner syndrome phenotypes. There is a blocking of the inactivation into the PAR regions which comprise the terminal 2.6 Mb of Xp in band p22.3; this segment has a homologous region on distal Yp. There is a secondary PAR that extends over 320Kb within distal Xq, having homology with distal Yq. An obligate recombination event occurs in the primary PAR of the X and Y chromosome at male meiosis; recombination between the secondary PAR, if it occurs at all, is infrequent. Certain other loci elsewhere on the X than in the PAR (some of which have homologs on the Y) are not subject to inactivation, and disomic expression of these genes in the female (and male) is normal. Lack of expression of these genes apparently account for the problems encountered in Turner syndrome.
Only one X per cell is active in any cell. Inactivation of "extra" X's arose to maintain the balance of genes between males and females (gene dosage compensation). Mammals do not tolerate extra chromosomes (plants, do) and X inactivation maintains the same number of active genes in the male and female. However, not all genes on the inactive X are inactivated, about 15% are still active. The inactive X is late replicating in the S phase of the cell cycle. It is hypermethylated and its histones are uniquely modified.
Barr discovered the Barr Body, the inactive X chromosome, as a darkly staining perinuclear body in the nuclei of female cat brains cells. Later, Mary Lyon, identified the Barr body as the inactive X chromosome, so the phenomenon of X inactivation is often called Lyonization. The Barr body is facultative heterochromatin. Inactivation occurs early in the embryo, at about the 20 cell stage. Inactivation is random, with a 50 - 50 chance of inactivating the maternal or paternal X. The mammalian female is a genetic mosaic with some of her cells with the XP active and some with the XM active. A woman with an X linked trait (e.g., Duchenne Muscular Dystrophy or anhidrotic ectodermal dysplasia) may manifest some of the features of the trait in those cells in which the mutant gene is expressed.
The role of the Y in mammalian male sex determination was first confirmed when a male calico cat was found. As mentioned earlier, ordinarily, calico cats are females since the genes for the gold and black colors they have are alleles on the X chromosome. Both alleles have to be present for the cat to be calico and so a calico cat needs two X chromosomes. The pattern of a calico cat is a perfect example of X inactivation pattern. The cat has a white background with more or less circular patches of of gold and black. Each area of color is the result of a clone of one cell with only the black or gold gene turned on. When a male calico cat was found, he had an XXY sex chromosome complement! (Alas, as in Klinefelter males, the cat was sterile.) Before this finding, it was not certain if it was the presence of two X's that determined femaleness and the lack of the second which produced maleness. In Drosophila, the fruit fly, which had contributed so much to the knowledge of genetics, females were XX and males were XO.
Females with translocations involving the X chromosome and autosomes will at first randomly inactivate the XP or the XM but if one of the resulting cell lines results in too much genetic imbalance, it will die out. The result may be an apparent skewing of the X inactivation pattern.
X chromosome inactivation occurs if the number of X chromosome exceeds one. Females with XX inactivate one X, males with XXY inactivate one X, triple X females (XXX) inactivate two X's. This is a mechanism of "dosage compensation" so that the mean amounts of gene products of X-encoded genes are the same in females and males. The Lyon hypothesis and later information about X chromosome inactivation has led to these conclusions: Normally each diploid cell has one active X chromosome, any "excess" (more than one) X's are inactivated. This is believed to have evolved as a dosage compensation mechanism between the sexes. The expression levels of the great majority of X-encoded genes are equalized between XY males and XX females by permanent silencing of one or the other X chromosome in the cells of female somatic tissues.
Inactivation takes place at the blastocyst stage (possibly at 10 - 20 cells) and is random for the paternal and maternal X chromosomes. However, in some tissues, especially the extra-embryonic membranes, the paternal X is preferentially inactivated. In marsupials, the paternal X is always inactivated. This phenomenon may be because the paternal X is inactivated during male meiosis and fails to get turned on again.
Some conditions appear to be exceptions to the rule that X inactivation is random. However, these "exceptions" are actually due to selection against imbalanced somatic cell lines. When a cell has one normal and one abnormal X, the abnormal chromosome is inactivated (provided it contains the inactivation center). In carriers of a balanced reciprocal X:autosome translocation, the normal X is inactivated. (Sometimes in a minority cell line, the translocation chromosome forms the Barr body.)
(a) is the 20-30 cell embryo with both X (maternal) and X (paternal) active
(b) the embryo in which random X inactivation has occurred resulting in a mosaic
(c) the embryo later when there has been selection against one of the two cell types Xm or Xp
(d) the embryo in which there is no selection and there are both Xm and Xp cells (mosaic)
In females with an unbalanced X:autosome translocation, the translocation chromosome is inactivated. Inactivation may or may not spread to the autosomal segment. (In some patients additional cell lines with different inactivation patterns have been found.) No exceptions seem to exist to the rule that inactivation is permanent in all descendants of a cell in somatic tissues--they form a clone. A female is normally a mosaic of two cell populations, each expressing gene alleles from either her paternal or her maternal X chromosome.
The tip of Xp (pseudo autosomal region) of the inactive X remains active; however, the level of activity of the genes in this region is less than that of the corresponding genes on the active X. One or two other regions on the inactive X (the Q-dark regions on both sides of the centromere) also remain active. There are four types of genes on the X "inactive" chromosome: those that are inactivated, those that escape inactivation, those that may or may not escape inactivation, and those that are reversible and can be off or on.
Attempts to de-repress the inactive X experimentally have met with limited success. This may, however, happen spontaneously in some malignant tumors or with aging.
No exceptions seem to exist to the rule that both (all) X chromosomes are active during meiosis in the oocytes.
The inactive X chromosome forms a condensed Barr body and is late-replicating during the S period.
The X and Y chromosomes are, without exception, inactivated in spermatocytes and apparently stay inactive through meiosis. One explanation for the sterility of males with an X:autosome translocation is that such a chromosome may be unable to undergo inactivation in male meiosis.
Sex Influenced, Sex Limited and Sex Linked Traits
Sex linked, sex influenced and sex limited traits should be distinguished from one another. Sex linked traits are those whose genes are on the X or Y chromosome. Sometimes because there are so few genes on the Y chromosome, they are referred to as X linked. Because of their location, these genes are inherited differently than genes on autosomes. Most of the genes on the X and Y chromosomes are not involved in sex determination although a few are and they are very important as we will discuss in a later lecture. Sex influenced and sex limited traits are, in general, coded for by genes on the autosomes (this makes sense since there are 22 autosomes and only two sex chromosomes). Sex limited traits are those that are expressed only in one sex or the other such as secondary sex characteristics (e.g., breasts, fat distribution, genitalia). Sex influenced traits are those that are found in both sexes but are inherited differently in the two sexes. Examples are baldness, voice range, and height. Baldness is dominant in males and recessive in females. Sex influenced traits are hormonally influenced (i.e., castration can prevent baldness).
Structural Chromosome Abnormalities
Chromosome structural changes include a wide variety of rearrangements including translocations, inversions, rings, isochromosomes some of which involve duplications or deletions of variable amounts of chromosome material. When apparently balanced rearrangement are found in an amniotic fluid culture the parents must be karyotyped to establish whether the fetus has a de novo rearrangement or one which is inherited from a normal parent. If inherited, they are usually harmless but if they are de novo, even apparently balanced structural rearrangements have a risk of causing congenital abnormalities. Some rearrangements where there is no apparent loss of genetic material would be expected to be innocuous in the bearer, however, they turn out to be a problem during meiosis. This type of problem arises with inversions and translocations that can form abnormal quadrivalents during crossing over and recombination in meiosis.
Structural chromosome abnormalities include breaks in chromosome arms. The most common abnormalities are terminal deletions of pieces from one end of a chromosome or interstitial deletions within an arm; inversions, either pericentric (including the centromere) which can change the shape of the chromosome or paracentric (in one arm) which will not; isochromosomes which are believed to result from mis division of the centromere to give a chromosome with 2 p arms or 2 q arms; ring chromosomes which result from two breaks, one in each arm, and a resealing into a ring; reciprocal translocations in which pieces of two chromosomes are exchanged; Robertsonian translocations, or dicentric fusions, usually involving the D and G group chromosomes. (A 21:21 balanced translocation carrier can never have a normal child.). When a structural chromosome abnormality is found in a fetus, the parents chromosomes should be karyotyped to see if they carry a balanced rearrangement or the same rearrangement or whether the abnormality in the fetus arose de novo. In general, de novo rearrangements carry a greater risk of abnormality than inherited ones.
Structural changes in the normal chromosome complement are compatible with normal development if there is no loss or gain of chromosome material. It is said to be "balanced." However, a structural change found in a fetus from an amniocentesis, for example, may appear to be balanced but could be missing some material. The parents are karyotyped and if one or the other has the same structural change the fetus will probably be normal. However, if the change is de novo there are risks of congenital abnormalities. Dorothy Warburton's 1991 article (Am. J. Hum Genet. 49:995-1013) is a valuable resource for genetic counselors when they give the risk figures for de novo chromosome rearrangements. She collected data on 377,357 reported amniocenteses over a 10 year period.

Types of structural chromosome changes
The Terminology for Structural Changes
Translocations (can be reciprocal or non-reciprocal; balanced or unbalanced)
t = translocation 45,XX,t(14;21)(p11;q11); this is a normal person with a balanced translocation between chromosomes 14 and 21. The breakpoints are given with the #14 first and the #21 last. Robertsonian translocations (centric fusions) often occur between the D and G group chromosomes. The human chromosome #2 is the result of a translocation of two ape chromosomes. Carriers of balanced translocations (who are normal) produce both balanced and unbalanced gametes with duplications and deletions of large pieces of the chromosomes involved. Their offspring with a duplication or deletion will have partial trisomy or monosomy.
Robertsonian translocations (centric fusions)
Translocations can be Robertsonian (centric fusion) reciprocal or merely the loss of material from one chromosome attached to a different chromosome. Translocations, too, can be inherited or de novo. Normal individuals can have balanced translocations. However, when they form gametes, they may not include the correct amount of chromosomal material. They can give too much or too little resulting in trisomy or monosomy of chromosomes or portions of chromosomes. Generally speaking, if the male is a translocation carrier, there is a lower risk of recurrence than if the female is the carrier. Most of the time people only become aware of being a translocation carrier after the birth of abnormal children and subsequently the karyotyping of the child and then themselves. Couples, where one is a carrier, can be offered amniocentesis and elective termination of chromosomally unbalanced fetuses in future pregnancies.
Robertsonian translocations or centric fusions occur among the D and G group chromosomes. The p arms of both are usually lost in the fusion. Sometimes there is a dicentric chromosome formed but it is unstable with two centromeres. The p arms of the D and G group chromosomes contain the highly repetitive rRNA genes so if there is a loss of two p arms in a translocation, there are still eight p arms left. In fact, some believe that the reason you see D and G group translocations so frequently is because of the homology between the p arm DNA. On the other hand, they are the only chromosomes we have that we can afford to lose p arms from because of the redundancy of genes there. When a translocation occurs between two 21 chromosomes the balanced translocation carriers can never have a normal offspring. They will either contribute no 21, which results in monosomy 21 and is lethal, or they contribute the 21/21 translocation which results in a Down syndrome child. Other Robertsonian translocation carriers with translocations involving chromosome 21, can have normal children, Down syndrome children, and more than the usual number of miscarriages. When a carrier parent gives the translocation chromosome and one of the normal homologs, the child will be trisomic and have uniparental disomy. Robertsonian translocations involving the same chromosome have a higher incidence of uniparental disomy. This means that both arms have come from the same parent chromosome.
Reciprocal and non-reciprocal translocations
Reciprocal translocations can occur between any two chromosomes. A piece of one is translocated to another. When this occurs, the carrier may have a balanced translocation. However, when meiosis occurs, the balanced translocation carrier will produce a variety of gametes some of which carry the normal homolog, some carry the balanced reciprocal translocation and some of which result in unbalanced gametes with duplications or deletions of the pieces of chromosome involved in the translocation. Geneticists refer to these conditions as partial trisomies and partial monosomies depending upon which combination the fetus receives. When a child has multiple congenital anomalies (MCA) one usually does a chromosome analysis. When a duplication or deficiency of a portion of a chromosome is found, it is wise to test the parents to see if they carry a translocation. If they do, amniocentesis should be offered in future pregnancies. If it is de novo, there is a negligible risk of recurrence.
Balanced reciprocal translocations differ from Robertsonian translocations. Gametes of balanced reciprocal translocation carriers can contain unbalanced gametes with deletions and duplications but do not result in trisomies and monosomies, only partial monosomy or trisomy.
Non reciprocal translocations also occur. The same information as for reciprocal translocations applies if they are inherited.
Cri du chat 5p-
![]()
Unbalanced Translocations: Left shows cytogenetic interpretation and right shows FISH corrected results
Cat-eye syndrome due to a duplication
Patients with the rare dysmorphic, highly variable syndrome known as the Cat-eye syndrome which includes coloboma (slit) of the iris and anal atresia (closed anus) have a "duplication" syndrome. They have four copies of a part of 22q [inv dup(22)(pterq11.2)].The extra copies are often in a supernumerary chromosome in which there is a duplication of part of the long arm of 22. The region that is duplicated can vary but there is a "critical region" responsible for the common phenotype. Most cases arise de novo, but familial (hereditary) transmission has been recorded including familial mosaicism. The phenotype appears not to correlate well with the size of the chromosome but it seems that four copies of the critical region is more likely to produce the phenotype than three copies.
i = isochromosome. These are chromosomes with two q or two p arms of the same chromosome. This could arise at anaphase if the centromere-kinetochore separates incorrectly, e.g., 46,X,i(Xq) is a Turner female with a normal X and an X made of two q arms.

Isochromosome
r = ring. A chromosome with a breakpoint in each arm can form a ring. This results in the deletion of varying amounts of the chromosome. Rings may be further modified or lost because of problems in mitosis. An example is 46,XY,r(4)(p15q34), with the breakpoints in each arm.
di = dicentric. These chromosomes have two centromeres and arise due to a translocation. They may experience difficulties at anaphase.
Inversions: pericentric and paracentric
inv = inversions may be paracentric (not involving the centromere) or pericentric containing the centromere). Inversions which do not have a breakpoint within a gene do not cause a problem in the carrier, however, in meiosis, one of the homologs must form a loop to pair up with the other. When loops are formed and recombination occurs within the loop, unbalanced gametes with duplications, deletions, dicentrics and acentrics result. Inversions are common differences between the chromosomes of humans and apes. Inversions are important in speciation since only those individuals with the same inversions can have normal meiosis and, therefore, normal offspring.
Some pericentric inversions are of no consequence An example of this is the inversion of the heteromorphic centromeric region of chromosome 9. The pericentric inversion, (inv)9(p11q13), is common and not usually associated with any problems. The larger the amount of material involved in an inversion, the greater the probability that during recombination, a loop will be formed so that the homologous regions can pair.
The risk of genetically grossly unbalanced gametes being produced in a person with a paracentric inversion is very high. In fact, the risk of them having an abnormal child is low since the abnormal gametes are so grossly imbalanced. Therefore, they will usually have a normal child (one who got a non recombinant chromosome) but one may see early fetal losses results from conceptuses from the gametes with the grossly unbalanced dicentric and acentric chromosomes.
On the other hand, pericentric inversions which involve breaks on both sides of the centromere, can produce gametes with chromosomes containing duplications and deficiencies due to crossing over within the loops. So carriers of pericentric inversions have a greater risk of live born abnormal children who are trisomic for some regions and/or monosomic for others.

Paracentric and Pericentric Inversions
del = deletion; dup = duplication. These can be due to unbalanced gametes from a translocation or inversion carrier. They result in partial monosomy or partial trisomy of the genes involved.
m = marker chromosome. These are small (unidentified) chromosomes with a centromere and may be of no consequence if they are "familial" but if they arise de novo in a fetus they may cause congenital anomalies. They are small, usually metacentric, fragments sometimes detected during routine karyotyping. Some familial ones arise in meiosis after a centric fusion between satellited chromosomes (50% involve 15 p which can be identified by DAPI + distamycin A staining). FISH probes can usually identify they origin of marker chromosomes. If the marker contains only repetitive and rDNA, there will be no clinical consequence. If other genes are included, there may be a problem.
pter = refers to the terminus of the p arm and qter = refers to the terminus of the q arm
der = derived; the abbreviation "der" is also used for a structurally rearranged chromosome. For example, 45,XY,der(14;21)(q10;q10) indicates a male with 45 chromosomes, in whom one normal chromosome 14 and one normal chromosome 21 have been replaced by a derivative chromosome arising from the translocation of the long arm (q) of chromosome 21 to the long arm of chromosome 14. This represents a Robertsonian translocation, or centric fusion.
mat = maternally derived; pat = paternally derived.
De novo balanced chromosome rearrangements and extra marker chromosomes carry risks for congenital abnormalities. The frequency of these de novo rearrangements and marker chromosomes and the risks for serious congenital anomalies associated with them were determined empirically by Dorothy Warburton. In summary the results were:
1/2,000 reported amniocenteses had a de novo reciprocal translocation; risk for serious congenital anomalies was 6.1%
1/9,000, reported amniocenteses had a de novo Robertsonian translocation; risk for serious congenital anomalies was 3.7%
1/10,000 reported amniocenteses had a de novo inversion; risk for serious congenital anomalies was 9.4 %
1/25,000 reported amniocenteses had an extra structurally abnormal chromosome of unidentifiable origin; risk was 14.7 % non satellited marker chromosomes and 10.9 % for satellited marker chromosomes.
Cri-du-chat is a deletion of 5 p (5p-); Wolf (or Wolf-Hirschhorn) is 4 p-. Patients with these syndromes (and others with deletion or duplication syndromes) often have parents with balanced translocations involving the chromosomes in which the child has the deletion or duplication.
Examples of Structural Chromosome Abnormalities
| KARYOTYPE | COMMENT |
| 46,XY,t(5;10)(p13;q25) | Balanced reciprocal translocation involving chromosomes 5 and 10 (break points indicated) |
| 45,XX,t(13;14)(p11;q11) | Centric fusion translocation of chromosomes 13 and 14. A Robertsonian translocation normal carrier |
| 46,XY,del(5)(p25) | Short arm deletion of 5, Cri du chat syndrome |
| 46,XX,dup(2)(p13p22) | Partial duplication of the short arm of chromosome 2 (p13p22) |
| 46,X,i(Xq) | Isochromosome of Xq; Turner female |
| 46,XY,r(3)(p26q29) | Ring chromosome 3 (p26q29) |
| 46,XY,inv(11)(p15q14) | Pericentric inversion of chromosome 11 |
Chromosome instability and repair defect syndromes.
There are several Mendelian disorders (AR or XR) which involve chromosome breakage are thought to be due to mutations in DNA replication or repair mechanisms. Many of them have disturbances of growth and development, defects in the immune system/bone marrow system, and all have a predisposition to malignancy. These include: 1. Bloom syndrome which exhibits sister chromatid exchange (SCE) in the cell cultures of those affected and is more frequent in Ashkenazi Jews. It is due to a defect in a DNA ligase. 2. Fanconi (anemia) syndrome with short stature, absent radii and hypoplastic thumbs, brown pigmentation, anemia, pancytopenia, greater risk for leukemia, it is diagnosed with a clastogen, diepoxybutane which induces broken chromosomes in the affected persons cultured cells. There is an increased sensitivity to alkylating agents. 3. Ataxia telangiectasia results in cerebellar ataxia and greater risks for malignancy even in the heterozygotes. Heterozygotes are 1.4% of the population and are found among those women with breast cancer is greater frequency. There is an increased sensitivity to radiation. Translocations involving chromosomes 7 and 14 are common in the cultured cells of these individuals. Cancer therapies using radiation (and chemo?) can be disastrous when used on these people when they have cancer. 4. Roberts syndrome shows limb reduction, mental retardation, severe growth deficiency. It is due to premature separation of centromeric heterochromatin in metaphase. 5. Cockayne syndrome is an autosomal recessive trait which shows growth failure, early developmental delay, progressive neurological dysfunction, and behavioral and intellectual deterioration. The defect is in DNA repair and can be diagnosed using skin fibroblasts. This syndrome shows locus heterogeneity and a range of phenotypes. Direct DNA testing is available on a research basis on the two of the genes known to be mutated in this disorder.
Xeroderma pigmentosum was one of the first disorders shown to be a due to defects in DNA repair. Specifically, it results in the inability to repair UV damage to DNA. Clinically, the patient has multiple skin cancers and corneal scarring. Some forms also affect the nervous system. The diagnosis can be made from cell cultures of affected individuals where the cells do not take up radioactive thymidine after being exposed to UV light. This is indicative of their inability to repair the pyrimidine dimers in the DNA which form due to UV exposure. It was found that when cells from two different people when grown together in culture took up the radioactive thymidine because they were able to correct the UV damage. When cultured cells from two different people corrected one another, they are said to be in different complementation groups. Sometimes two patient's cells did not correct one another, thus they were in the same complementation group. At least 9 complementation groups have been found and the interpretation is that mutations in at least 9 different genes (and gene products) can cause this disorder. This should not be surprising since it is known that the repair pathway involves several steps and several (multimeric) enzymes are involved. This is an example of a specific type of genetic heterogeneity (genocopies) known as locus heterogeneity. We were already familiar with genetic heterogeneity (but not locus heterogeneity) when we talked about Down syndrome being due to straight trisomy 21 or translocation Down or partial duplications of parts of 22q. Locus heterogeneity refers to the situation whereby the same or clinically similar genetic disorders can arise from mutations in totally separate genes. There are many examples of this in human genetics. Another way of expressing this situation is that the same phenotype can be due to different genotypes.
Addenda
Here are two exchanges of E-mails from and to parents of children with chromosome abnormalities. These parents had found this web site and were hoping I could give them more information than they had. Alas, that is not easy with partial deletions since no two are alike. Here are my letters. The first one is from a parent with a child with an interstitial deletion of 2q12-14.1 and the second child has a partial trisomy of chromosome 16p.
Parent #1 "I'm not even sure how, but I stumbled upon your website in my continuous efforts to gain more knowledge about my 17 month old son's recent diagnosis. After watching our son fail to meet major milestones (and still not meeting them), we ultimately put him through genetic testing. The results are that he has an interstitial deletion of 2q12-14.1. We have already met with a geneticist and her prognosis is not very optimistic. I'm wondering if you might have any resources for me to consult and/or if you've had experience with this particular deletion. Its my understanding that there is no other reported case with his specific deletion and there are only 4 others with 'similar' deletions. Any information or resources you could provide would be greatly appreciated.
Response: "Rena Falk, the clinical genetist you saw, is an expert in this field and one to whom we turn for second opinions so you are in good hands. Since, you have had the explanation from Dr. Falk and also you have done a lot of research on your own, you probably know as much as I can tell you. If you haven't seen the web site, http://www.chromodisorder.org/ you might want to look at it. It may not be very helpful with your specific situation, however.
I did a search in the Online Biomed library at UCLA and found a reference to an article: "Interstitial deletion of the long arm of chromosome 2: a clinically recognizable microdeletion syndrome, Clinical Dysmorphology, 2000" However, the Clinical Dysmorphology journal Online begins in 2001 and I cannot access the 2000 issue. You could go to the UCLA Biomed library and get it however, since they will have the hard copy. The Biomed library is on the first floor of the UCLA Medical School. The reference librarian can help you find the journal so you can copy the article.
It is good that you and your other son are being tested. You or your husband could have what we call a "balanced translocation" of the 2q12q14.1 region missing in Conner. If so we would see the deletion in one of your chromosomes 2 and the piece would be somewhere else.....on another one of your chromosome. These microdeletions are not uncommon but many result in miscarriages so they are not detected. Microdeletions, although called "micro" involve large amounts of DNA and therefore tens to hundreds of genes. Deletions, as you might suspect, are generally felt to be more deleterious that additions of extra material....although both cause serious problems. For example, Down syndrome is due to an extra chromosome 21 (the chromosome with the least number of genes) but having only one chromosome 21 (deletion of an entire chromosome, known as monosomy) is lethal in utero. Conner has a partial monosomy of chromosome 2.
These deletions can be inherited via a "carrier" parent but the question still remains about what causes them originally. We know that certain mutagens such as ionizing radiation can cause deletions, another known mechanism is abnormal "crossing-over" and exchange in meiosis....the cell division that gives rise to eggs and sperm. Normally your maternal and paternal chromosomes pair up during meiosis (in your ovary or testes), the arms of the chromosomes cross over, a break occurs in each with resealing and the exchange results in new combinations of genes in the chromosomes which then go to the gametes (eggs and sperm). However, sometimes there is a mismatch and the two (homologous) chromosomes do not pair up as they should and when the subsequent breakage and reunion occur there may be additional DNA on one chromosome and less DNA (microdeletion) on the other. These type of mistakes are much more common than people might think. Until we had better ways of looking at chromosomes many cases of micro additions and microdeletions went undiagnosed as to underlying cause."
Parent 2: Hi I am a mom of a child with a duplication of the short arm of chromosome 16, I saw your website and was wondering if you have anything on this disorder. she is 8 years old.
Response: There is an excellent web site at: http://www.trisomy16.org/ which describes a variety of chromosome 16 rearrangements. The section on partial trisomy 16 is the most appropriate for your daughter. As I am certain you know by now, the exact expression and symptoms of the duplication cannot be predicted because each child has a different genetic background....they have inherited many other genes from their parents beside the extra ones on chromosome 16. Also, these duplications contain hundreds to thousands of genes and they are not the same in each child. However, you might find the child called Lauren interesting. She is under the partial trisomy 16 page of the web site: http://www.trisomy16.org/html/partial.html#lauren
You did not say but I assume this duplication was found in all of her cells. Also were you and her father karyotyped? Occasionally, one of the parents is a "carrier" of a balanced translocation. I assume you have seen a clinical geneticist and s/he has explained this to you. If not tell me where you live and I will put you in contact with one.
Parent 2 response: That is funny you mentioned the website I am already very involved, my daughter is on it xxxxxxxx not too far under lauren (who by the way is a great kid I met her last year at the world congress conference). I was just hoping and always looking, I am putting together a conference here on Long Island along with others from the foundation. If you are interested, it is July16-20 of this year. (Neither) my husband nor I are carriers and I have two healthy older children (17 & 14). Thank you for our response and time,
